journal menu
journal menu





issue contents
February 2010 issue
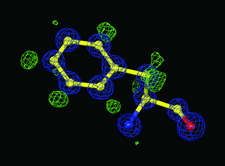
Cover illustration: The 0.98 Å resolution crystal structure of the 210 kDa protein endoNF in complex with sialic acid is presented in a new crystal form (p. 176).
editorial




Free 

research papers


Crystal structures of free and ATP-bound forms of Ap4A hydrolase from A. aeolicus have been determined at 1.8 and 1.95 Å resolution, respectively. The ATP-bound crystal structure shows binding of the product molecule at the primary active site and of the substrate molecule at a noncatalytic site near the N-terminal region of the enzyme.
Open access
access
The paper describes the software package XDS for processing of single crystal diffraction data recorded by the rotation method.
Open access
access
The working principles of important steps in processing rotation data are described as employed by the program XDS.

The structure of human apoptosis-related protein kinase MST3 in its active form was determined with various bound ligands including Mn-ADP.

The crystal structure of purine nucleoside phosphorylase from grouper iridovirus was solved at 2.38 Å resolution.
PDB reference: grouper iridovirus PNP, 3khs
Open access
access
P212121 crystals of SIV Nef core domain bound to a peptide fragment of the T-cell receptor ζ subunit exhibited noncrystallographic symmetry and nearly perfect pseudo-merohedral twinning simulating tetragonal symmetry. For a different peptide fragment, nontwinned tetragonal crystals were observed but diffracted to lower resolution. The structure was determined after assignment of the top molecular-replacement solutions to various twin or NCS domains followed by refinement under the appropriate twin law.

The 0.98 Å resolution crystal structure of the 210 kDa protein endosialidase NF in complex with sialic acid is presented in a new polymorphic form.
PDB reference: endosialidase NF, 3ju4
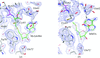
The crystal structures of activated S-adenosylmethionine decarboxylase (AdoMetDC) from T. maritima and of its complexes with S-adenosylmethionine methyl ester and 5′-deoxy-5′-dimethylthioadenosine have been obtained. Comparison of the structures with that of human AdoMetDC provides insights into the substrate specificity, binding mode, autoprocessing and evolution of the enzyme.
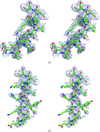
The structure of human desArg-C5a has been determined using molecular replacement to 2.58 Å. The structure is a dimer, and the N-terminal helices in each monomer differ from each other and occupy completely different positions from the NMR structures.
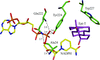
The structure of the Y224D mutant of mouse 3(17)α-hydroxysteroid dehydrogenase revealed that the mutation resulted in a change in the conformation of the flexible loop B. This loop is a unique feature of the active-site architecture of the wild type and is formed by the side chains of Tyr224 and Trp227.
PDB reference: Y224D mutant AKR1C21, 3fjn

This first structure of a fungal ortholog of ASA dehydrogenase has a similar overall fold and domain organization to the other members of this enzyme family. However, differences in the binding of the nucleotide cofactor and changes in the nature of the subunit interface are proposed to account for the lower catalytic efficiency of this enzyme from C. albicans.
PDB reference: caASADH, 3hsk
Open access
access
The PHENIX software for macromolecular structure determination is described.
addenda and errata
Free

Retraction of two articles by H. M. Krishna Murthy et al..