

 journal menu
journal menu





issue contents
August 2012 issue
Cover illustration: Crystal structure of an alternating D-alanyl, L-homoalanyl peptide nucleic acid (PNA). The molecule forms a tetrameric cage with standard Watson-Crick base pair geometry between the monomers (p. 1067).
research papers






The three-dimensional structure of an industrially relevant cellulase in complex with ligands has been determined at a resolution of 1.9 Å.
PDB reference: CtCel6A, 4a05

A low-resolution structure of the catalytic subunit CK2α of human protein kinase CK2 (formerly known as casein kinase 2) in complex with the ATP-competitive inhibitor resorufin is presented. An overview of all published human CK2α crystal packings serves as the basis for a discussion of the factors that determine whether the open or the closed hinge/helix αD conformation is adopted.

Open  access
access
access
Acoustic droplet ejection achieves precise, tipless, non-invasive transfer of diverse aqueous solutions, enabling nanolitre-scale crystallization trials. The rapid and scalable technique demonstrated successful crystal growth with diverse targets in drop volumes as small as 20 nl.
A mutant variant of the Synechococcus elongatus PII protein (PII-I86N) has been identified to have impaired 2-oxoglutarate binding. Here, the PII-I86N variant was cocrystallized in the presence of ATP, magnesium and citrate and its structure was solved at a resolution of 1.05 Å.
PDB reference: PII-I86N, complex with ATP, magnesium and citrate, 4aff
O-Acetylserine sulfhydrylase (OASS) from L. donovani can bind to serine acetyltransferase (SAT) C-terminus mimicking peptides with a range of bulkiness; the strength of these interactions correlates with the size of the peptide side chains and the size of the active-site cleft. Differences in the interactions of OASS and SAT from various species may be explained by the size of the active-site cleft.
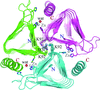
Crystal structures of YrdA from E. coli show conformations that have not been reported for γ-class carbonic anhydrase family proteins.

The structure of ustiloxin D bound to tubulin was determined at a resolution which allows interatomic interactions to be defined and provides a basis for the structure-based design of ligands with improved activity. It is also shown how local interactions of vinca-domain ligands with tubulin influence the relative positioning of the latter and lead to the large-scale polymorphism of ligand-mediated tubulin assemblies.
Open access
access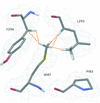
All-atom models derived from moderate-resolution protein crystal structures contain a high frequency of close nonbonded contacts, independent of the major refinement program used for structure determination. All-atom refinement with PrimeX corrects many of these problematic interactions, producing models that are better suited for use in computational chemistry and related applications.
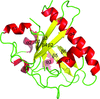
The structure of the C-terminal domain of yCdc73 revealed a GTPase-like fold.
PDB reference: conserved domain of yeast Cdc73, 4dm4
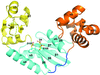
The structure of a Y-family DNA polymerase from Mycobacteria named MsDpo4 along with solution studies suggest that the PAD region is endowed with the ability to adopt multiple orientations. This attribute would allow the enzyme to accommodate alterations in the width of the DNA double helix to facilitate its twin functions of trans-lesion bypass and adaptive mutagenesis.
PDB reference: MsPolIV, 4dez
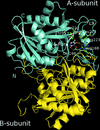
X-ray structures and in silico models are reported for complexes of the drug 5-fluorouracil with uridine phosphorylases from S. typhimurium and V. cholerae.
PDB reference: StUPh–5-fluorouracil, 3nsr
Open access
access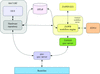
A powerful and easy-to-use workflow environment has been developed at the ESRF for combining experiment control with online data analysis on synchrotron beamlines. This tool provides the possibility of automating complex experiments without the need for expertise in instrumentation control and programming, but rather by accessing defined beamline services.
Open access
access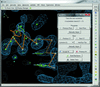
RCrane is a new tool for the partially automated building of RNA crystallographic models into electron-density maps of low or intermediate resolution. This tool helps crystallographers to place phosphates and bases into electron density and then automatically predicts and builds the detailed all-atom structure of the traced nucleotides.

The crystal structure of the branched-chain aminotransferase from S. mutans is reported at 1.9 Å resolution.
PDB reference: SmIlvE, 4dqn

A rapid plate-based pH assay has been developed that takes advantage of the automation available in a protein-crystallization laboratory.
The amino acid L-proline is shown to be a good cryoprotectant for protein crystals. Four examples are provided; the range of proline used for cryoprotection is 2.0–3.0 M.
Open access
access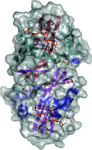
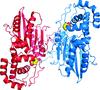
Glucosamine-6-phosphate N-acetyltransferase is an essential enzyme of the eukaryotic UDP-GlcNAc biosynthetic pathway. A crystal structure at 1.55 Å resolution revealed a highly unusual covalent product complex and biochemical studies investigated the function of a fully conserved active-site cysteine.
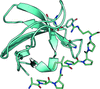
The promiscuous binding of an NS5A peptide to the Fyn SH3 domain is described.

The first X-ray structure of p38α cocrystallized with TAK-715 is presented, as well as new high-resolution structures of p38α bound to SB-203580, SCIO-469 and VX-745. The impact of crystallization conditions and selectivity profiles on protein conformation is discussed.
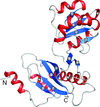
The crystal structure of the isolated full-length ribosomal L1 stalk has been solved at high resolution, detailed analysis of L1-rRNA interactions and crystal packing has been carried out.
PDB reference: ribosomal L1 stalk, 3u4m
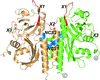
Crystallization of RpBphP2-CBD was achieved by homologue-directed mutagenesis via dimer formation in solution by the mutation N136R. Naturally occurring mutations in BphPs correlate well with their observed dimerization properties and suggests a structural diversity within this family.
PDB reference: RpBphP2-CBD*, 4e04
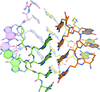
The 1.0 Å crystal structure of an alternating L-homoalanyl, D-alanyl peptide nucleic acid reveals a novel tetrameric cage with Watson–Crick pairing of the stacked nucleobases.
PDB reference: PNA1, 3c1p





