journal menu
journal menu





issue contents
February 2013 issue
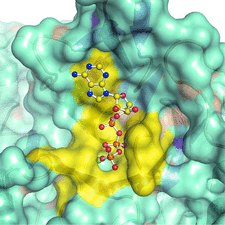
Cover illustration: The arrangement of ATP, Mg2+ and key amino-acid residues in the active sites present in T. tengcongensis DppD (p. 256) as a cartoon representation and surface. The residues coloured yellow interact with ATP and Mg2+ and form the binding pocket.
editorial




Free 

research papers


The crystal structure of the the natural fusion protein EpsAB from V. vulnificus is described.
PDB reference: EpsAB, 4g54

The potential causes of severe misinterpretation of ligand electron density in a surprisingly large number of protein–ligand structure complexes are analyzed. Techniques and tools are presented together with suggestions for good practice aimed at minimizing overinterpretation and mistakes in protein–ligand structure models.

The full-length crystal structure of Toxascaris leonine galectin (Tl-gal), a galectin-9 homologue protein, was determined at a resolution of 2.0 Å. The NCRD of Tl-gal resembles that of human galectin-7 and its CCRD resembles human galectin-9, but the residues in the interface and loop regions of the NCRD and CCRD are flexible and are related to interaction.
PDB reference: Tl-galectin, 4hl0
Open access
access
The statistical effects of translational noncrystallographic symmetry can be characterized by maximizing parameters describing the noncrystallographic symmetry in a likelihood function, thereby unmasking the competing statistical effects of twinning.
A 2.15 Å resolution crystal structure of TM0159 with bound IMP and enzyme-kinetic data are presented. This noncanonical nucleoside triphosphatase from T. maritima helps to maintain a correct pool of DNA and RNA precursor molecules.
PDB reference: TM0159, 3s86

The results observed for three different crystal forms of the enzyme PurE are reported and an attempt is made to infer some initial conclusions regarding the role that the solvent, the crystallization agents (mainly salts and polyethylene glycol) and interactions within the protein structures play in determining the changes in the diffraction pattern upon alteration of the external relative humidity of the crystals.
The structure, reaction mechanism and behaviour of a promiscuous plant nuclease are described. Structure solution was achieved by a combination of MAD and MR on multiple crystal forms.

The crystal structure of B. subtilis RibG complexed with a deaminase product was determined and the yeast Rib2 structure was modelled. Subsequent mutational analysis was carried out to illustrate its distinct substrate specificity. An essential amino-binding hole which is conserved in the RNA/DNA-editing deaminases was also identified.
PDB reference: RibG, 4g3m
The crystal structure of the C-terminal half of human XPB helicase was determined to 1.8 Å resolution. A disease-causing mutation (XP11BE) at the C-terminus produces a less soluble XPB mutant, significantly reducing the cellular level of the TFIIH complex.
PDB reference: C-terminal half of human XPB helicase, 4ern
High-resolution crystal structures of the oncoprotein Rcl in complexes with apoptotic nucleotides have been solved by molecular replacement using combined information from NMR, SAXS and fold recognition.
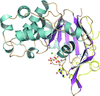
The crystal structure of DppD, the nucleotide-binding domain of a dipeptide ABC transporter from T. tengcongensis, bound with an iron–sulfur cluster is reported at 2.89 Å resolution.
PDB reference: DppD, 4fwi
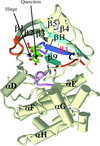
The crystal structure of quercitrin, a naturally occurring flavonol glycoside, has been determined in a complex with the N-terminal kinase domain of murine RSK2. The structure revealed that quercitrin inhibits the RSK2 kinase in the same fashion as another known inhibitor, SL0101.

The structure of a polysaccharide deacetylase from B. cereus has been determined using an adventurous molecular-replacement procedure.
PDB reference: polysaccharide deacetylase, 4hd5
Open access
access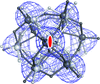
A new approach is presented that allows the efficient localization and orientation of heavy-atom cluster compounds used in experimental phasing by a molecular replacement procedure. This permits the calculation of meaningful phases up to the highest resolution of the diffraction data.
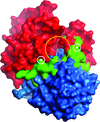
X-ray structures of Rhizobium sp. sucrose isomerase mutants have improved the understanding of the enzyme mechanism and have allowed the factors that govern the hydrolytic activity of this industrially important enzyme to be pinpointed.
short communications

High-pressure-freezing allowed low-temperature data collection from a single virus crystal for which no standard cryoprotection could be established.