

 journal menu
journal menu





issue contents
January 2014 issue
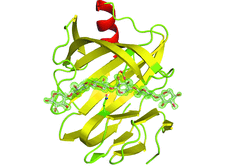
Cover illustration: An E177Q variant of the family 11 xylanase secreted by the filamentous fungus Trichoderma reesei complexed with xylohexaose (p. 11).
letters to the editor




Free 

Recent examples of the misrepresentation of biological helical structures in the popular media are identified and discussed.
research papers


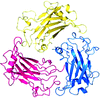
TRAF4 is a unique family of TRAF, which participates in many ontogenic processes. The crystal structure of TRAF domain of TRAF4 was solved at 2.3 Å resolution.
PDB reference: human TRAF4 TRAF domain, 4k8u
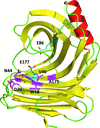
X-ray structures of a family 11 xylanase with bound xylohexaose substrate or products revealed six sugar-binding subsites in the active site of the enzyme. Replacing amino acids near the catalytic glutamic acid 177 decreased the xylanase activity but increased the relative activity at alkaline pH.
PDB references: E177Q–X6, 4hk8; N44H–X3, 4hk9; ligand-free N44H, 4hkl; XynII–TrisX2–X3, 4hkw; ligand-free E177Q, 4hko
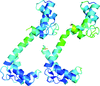
Novel bent conformations of the calmodulin central helix are presented. The structures may relate to the initial stages of structural collapse in calmodulin upon target-peptide binding.

An analysis of the rotational order–disorder structure of the reversibly photoswitchable red fluorescent protein rsTagRFP is presented.
PDB reference: rsTagRFP, 4kpi
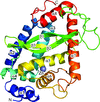
The crystal structure of NleC is reported at 1.55 Å resolution. In conjunction with biochemical analyses, the structure reveals that NleC is a member of the zincin zinc protease family and that the configuration of the NleC active site resembles that of the metzincin clan of metallopeptidases.
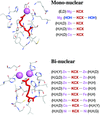
A computational method for the prediction of lysine carboxylation (KCX) in protein structures is described. The method accurately identifies misreported KCXs and predicts previously unknown KCX sites.
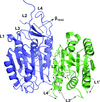
The regulatory mechanism of the caspase 6 pro-domain is revealed.
PDB reference: proCASP6H121A, 4iyr
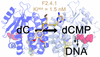
This article describes the structural and biochemical characterization of a new class of high-affinity and selective human deoxycytidine kinase inhibitors.

This work reports the crystal structure of BsrV, the broad spectrum racemase of Vibrio cholerae (1.15 Å resolution); unveils signature amino acids associated to its multispecificity and uses this signature sequence to identify a large family of BsrV-like multi-specific racemases in bacteria. Structural analyses of an additional Bsr confirmed the distinguishing features are conserved among Bsr-family members.
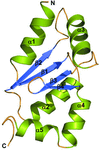
The structure of glutaredoxin 1 from P. falciparum has been determined at atomic resolution and its unique structural features have been dissected.

In serial crystallography, it is demonstrated that the indexing mode of partial data sets can be established using correlation coefficients against other data sets and a clustering procedure. For 24 chiral space groups clustering can be performed in two dimensions, but in space groups P3, P31 and P32 clustering in three or four dimensions is required.
Open access
access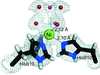
The level of structural detail around the metal sites in Ni2+ and Cu2+ T6 insulin derivatives was significantly improved by using a combination of single-crystal X-ray crystallography and X-ray absorption spectroscopy. Photoreduction and subsequent radiation damage of the Cu2+ sites in Cu insulin was followed by XANES spectroscopy.
Open access
access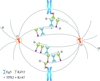
The structural and biochemical study of Kif15 provides insight into this potential drug target and allows comparison with Eg5, a kinesin that partially shares the functions of Kif15.
PDB reference: Kif1519–375, 4bn2
Open access
access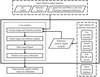
A new module, Guided Ligand Replacement (GLR), has been developed in Phenix to increase the ease and success rate of ligand placement when prior protein-ligand complexes are available.
Open access
access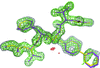
A software system for automated protein–ligand crystallography has been implemented in the Phenix suite. This significantly reduces the manual effort required in high-throughput crystallographic studies.
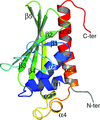
The FP domain of the human F-box protein Fbxo7 adopts an α/β-fold structure that is similar to the FP domain of the human proteasome inhibitor PI31. However, the two FP domains show dramatic differences in the way that they mediate protein–protein interactions.
Open access
access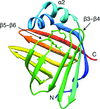
The structure of the human myelin peripheral membrane protein P2 has been refined at 0.93 Å resolution. In combination with functional experiments in vitro, in vivo and in silico, the fine details of the structure–function relationships in P2 are emerging.
PDB reference: human myelin peripheral membrane protein P2, 4bvm
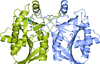
Crystal structures of the H. pylori methylthioadenosine nucleosidase (MTAN) HpyMqnB have been solved in native and complexed forms, shedding light on substrate binding and the reaction mechanism.
Open access
access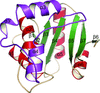
The crystal structure of a bacterial acetyltransferase with 27% sequence identity to the C-terminal domain of human O-GlcNAcase has been solved at 1.5 Å resolution. This S. sviceus protein is compared with known GCN5-related acetyltransferases, adding to the diversity observed in this superfamily.
PDB reference: SsAT, 4bmh
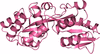
Crystal structures of FbpA from T. thermophilus (TtFbpA) in its apo and ferric ion-bound forms have been solved. TtFbpA shows the novel formation of a six-coordinated complex of ferric ion coordinated by three tyrosine residues, two monodentate bicarbonates and a water molecule.
short communications
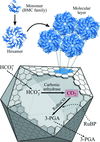
The authors describe the structure determination of a hexagonally layered protein structure that suffered from a complicated combination of translational non-crystallographic symmetry and hemihedral twinning. This case serves as a reminder that broken crystallographic symmetry resulting from doubling of a unit-cell axis often requires a new choice of origin.
PDB reference: CcmK1 shell protein mutant, 4liw





