journal menu
journal menu




issue contents
July 2014 issue
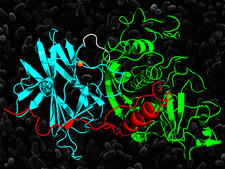
Cover illustration: The crystal structure of the cysteine protease and lectin-like domains of Cwp84, a surface layer associated protein of Clostridium difficile (p. 1983)
research papers





An ultrahigh-resolution structure of the Z-DNA dodecamer, solved from the anomalous signal of P atoms, reveals substantial flexibility of the backbone phosphate groups.
PDB reference: d(CGCGCGCGCGCG)2, 4ocb
Open  access
access
access
Structure–function studies of sialic acid-binding proteins from F. nucleatum, P. multocida, V. cholerae and H. influenzae reveal a conserved network of hydrogen bonds involved in conformational change on ligand binding.
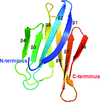
The structures of N-terminal and C-terminal domains of the human Fas apoptosis inhibitory molecule were determined. Structural and biochemical analyses of the two domains linked the interdomain interaction to the anti-apoptotic activity.

Download citation


Download citation
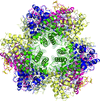
The structure of the deoxygenated form of the 400 kDa haemoglobin from the tube worm L. satsuma is reported.
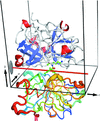
Crystal structures of wild-type L. enzymogenes LysC and of the K30R variant near the substrate-binding site have been determined in complex with the covalent inhibitor TLCK that mimics a tetrahedral reaction intermediate. H-atom density indicates a hydroxyl group in the oxyanion hole.
Open access
access
A novel procedure for identifying ligands in macromolecular crystallographic electron-density maps is introduced. Density clusters in such maps can be rapidly attributed to one of 82 different ligands in an automated manner.

Three crystal structures of potassium-dependent plant L-asparaginase were solved in complexes with K+, with Na+ and with both cations. A novel alkali metal-binding loop Val111–Ser118 (the activation loop) changes its conformation upon K+/Na+ exchange, leading to a reconfiguration of three key residues, His117, Arg224 and Glu250 (the catalytic switch), to allow or prevent substrate binding in the active site of the enzyme.
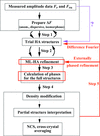
Small differences between heavy-atom derivatives can be exploited to improve the experimental phasing by treating each derivative as an independent SAD data set and combining these data sets with circular permutable pairs of SIR data sets. The basis for this generally applicable approach is that the effective resolution of isomorphous signals between highly isomorphous derivatives is often much higher than the effective resolution of the anomalous signals of individual derivative data sets.
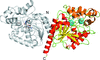
Two halide-binding sites were identified in the crystal structure of the newly isolated haloalkane dehalogenase DbeA from Bradyrhizobium elkanii USDA94. Elimination of the second halide-binding site significantly modified the enzyme substrate specificity, catalytic activity and stability in the presence of chloride salts. A shift in the substrate-specificity class after mutagenesis was demonstrated for the first time for haloalkane dehalogenases.
PDB reference: DbeA, 4k2a
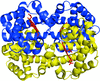
The structure of liganded cat Hb adopts a T-state-like quaternary structure owing to its ligand binding properties, which includes low cooperativity, ligand affinity and the blunt response to an allosteric effector.
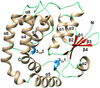
The structure of glutaredoxin 2 (Grx2) from Escherichia coli co-crystallized with glutathione (GSH) at 1.60 Å resolution is reported.
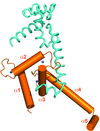
Crystal contacts in the structure of a chimeric protein composed of residues 1723–1818 of p300 Taz2 and residues 37–61 of C/EBP∊ reveal a possible mode of interactions of C/EBP transcriptional activators with p300/CBP.
PDB reference: C/EBP∊-p300 Taz2 chimera, 3t92
Open access
access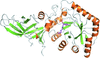
Structures of C. difficile alanine racemase in complex with the PLP cofactor and cycloserine are presented.
Open access
access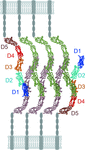
Crystal structures of Intercellular Cell Adhesion Molecule-5 (ICAM-5) show charge-based homotypic interactions and a potential ICAM-5 cell adhesion complex in neurons.
Open access
access
The heterodimeric structure of the MST1 and RASSF5 SARAH domains is presented. A comparison of homodimeric and heterodimeric interactions provides a structural basis for the preferential association of the SARAH heterodimer.
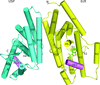
The crystal structure of the heterodimeric ligand-binding domains of the B. ovis ecdysone receptor was solved in the presence of an ecdysteroid and a synthetic methylene lactam insecticide, respectively. Analysis of these structures reveals a new level of conformational flexibility for the ecdysone receptor upon binding of ligands.

Elucidation of the crystal structure of the PPARγ F360L–LT175 complex provides considerable insights into the structural basis of the transactivation deficiency of this mutant, which has been associated with familial partial lipodystrophy.
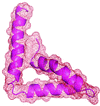
A new molecular replacement approach is reported utilizing a molecular envelope derived from NMR structures at low resolution.
Open access
access
The crystal structure of Cwp84, an S-layer protein from Clostridium difficile is presented for the first time. The cathepsin L-like fold of cysteine protease domain, a newly observed `lectin-like' domain and several other features are described.
PDB reference: Cwp84, 4ci7
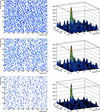
A variety of direct and reciprocal-space techniques have been combined for ab initio phasing of proteins at non-atomic resolution.
Open access
access
Pyrin domains (PYDs) recruit downstream effector molecules in NLR signalling. A specific charge-relay system suggests a the formation of a signalling complex involving a PYD dimer.
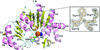
The crystal structures of PAP phosphatase-1 from E. histolytica and its complex with AMP have been determined at 2.1 and 2.6 Å resolution, respectively. This enzyme is a Li+-sensitive Mg2+-dependent inositol polyphosphate 1-phosphatase that catalyses the hydrolysis of both 3′-phosphoadenosine 5′-phosphate and inositol 1,4-bisphosphate.
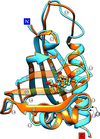
Two plant cytokinin-specific binding proteins were crystallized in complex with gibberellic acid (GA3), which is an entirely different plant hormone. The crystal structures, determined at high resolution, reveal a highly specific mode of GA3 binding, calling for a revision of the hormone specificity of plant proteins with the PR-10 fold.
Download citation
Download citation
Open access
access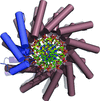
Crystal structures of BurrH and the BurrH–DNA complex are reported.
addenda and errata
Free

A correction is made to the article by Yoon et al. [(2013). D69, 1867–1875].