journal menu
journal menu




issue contents
April 2016 issue
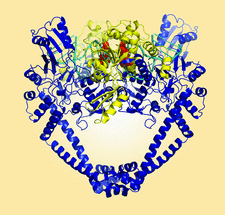
Cover illustration: The crystal structure of a Klebsiella pneumoniae topoisomerase IV ParC/ParE cleavage complex with DNA stabilized by levofloxacin (Veselkov et al., p. 488). The ParC subunit of the topoisomerase is shown in blue, the ParE subunit in yellow and DNA in cyan. Bound molecules of levofloxacin (a fluoroquinolone antibiotic) are shown in red. K. pneumoniae is a Gram-negative bacterium that is responsible for a range of common life-threatening infections and certain strains of Klebsiella have become highly resistant to antibiotics. In order to overcome this resistance, new drugs targeted against topoisomerase IV are being developed. The structure of the ParC/ParE-DNA-levofloxacin binding site highlights details that are essential for the rational design of Klebsiella topoisomerase inhibitors.
research papers








 access
access
 access
access access
access

 access
access
 access
access access
access access
access
book reviews