journal menu
journal menu





issue contents
November 2000 issue
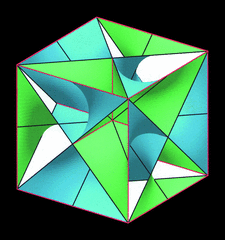
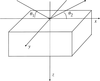
Cover illustration: Unit cell of a self-intersecting minimal surface with symmetry I432. The surface subdivides R3 into two congruent three-periodic mutually interpenetrating labyrinths with symmetry P4232. Lines of self-intersection are marked in red. See Koch [Acta Cryst. (2000), A56, 15-23].
research papers




A kinematic type of expression is derived for the dynamical diffracted-beam amplitudes and its validity is illustrated for thin films of GaAs and Au.

Temperature-dependent Debye–Waller B factors have been obtained for five compounds with the caesium chloride structure.

The phase problem is formulated as the minimization of a cost function with respect to the atomic coordinates. The cost function involves both the magnitude and phase of the calculated structure factors. Structures undergoing trial calculations include hexadecaisoleucinomycin.

False low-resolution crystallographic phases for a macromolecule generated under globular density constraints can be corrected via error-correcting codes.
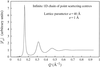
A model involving only a small number of parameters provides a convenient way of interpreting diffraction patterns from MCM-41 materials. Each parameter of the model has a clear physical meaning, and this approach is clearly superior to extracting pore structure information by fitting Gaussians to an observed diffraction pattern.
The statistical kinematical X-ray diffraction theory is developed to describe reciprocal-space maps from deformed crystals with structural defects.
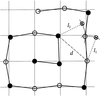
A simple model of the crystallite size–disorder relationship for paracrystalline materials (the 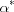 rule) based on the spiral paracrystal is described. Simulation results based on this model are presented.
rule) based on the spiral paracrystal is described. Simulation results based on this model are presented.
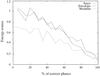
Simulated-annealing Monte Carlo methods in the multistart mode were successful in overcoming the phase-ambiguity problem in space group 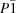 ; for the small protein rubredoxin. This involved minimization of an energy function derived from the Sayre equation and monitoring the Terwilliger criterion characterizing the roughness of the resulting maps.
; for the small protein rubredoxin. This involved minimization of an energy function derived from the Sayre equation and monitoring the Terwilliger criterion characterizing the roughness of the resulting maps.
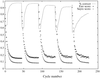
Mean-field optimization has been used to recast in a single formalism the problem of phase optimization using an arbitrary energy function in the presence of an experimentally determined phase probability function; this leads to the definition of a generalized figure of merit. Thermodynamic considerations lead to a new discussion of how to determine the relative weights in phase refinement and suggest that the free energy might be a better refinement target than maximum likelihood.

Topological analyses and space partitioning were performed on high-energy synchrotron-radiation data from Cu2O.

A new module interfaced to the XD programming package has been used in the evaluation of intermolecular interactions and lattice energies of crystals of p-nitroaniline, L-asparagine monohydrate and the pentapeptide Boc-Gln–Iva–Hyp–Ala–Phol. Comparison with ab initio supermolecular calculations at the HF, DFT and MP2 levels and with HF and DFT periodic crystal calculations shows agreement and discrepancies, the nature of which is discussed.
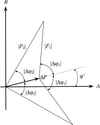
The direct method of breaking phase ambiguity is applied in solving noncentrosymmetric crystal structures from two-wavelength powder diffraction data.

Sampling the diffraction pattern of a finite specimen at a spacing finer than the Nyquist spacing (the inverse of the size of the diffracting specimen) corresponds to surrounding the electron density of the specimen with a no-density region. This no-density region can be used to retrieve the phase information directly from the diffraction pattern of (a) a small perfect or imperfect crystal or (b) a repeated motif without orientational regularity or (c) an unrepeated motif, such as an amorphous glass, a single molecule or a single biological cell.
short communications
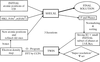
Specific problems in disordered supramolecular structures are solved by a new development of the TWIN algorithm, through the calculation of the `super-resolution wave function'.
international union of crystallography
Free 

Free