journal menu
journal menu





issue contents
January 2006 issue
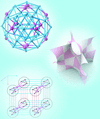
Cover illustration: Determination of a one-dimensional structure by the charge flipping algorithm. Bottom curves: grey is the initial electron density obtained with a random choice of structure-factor phases; white is the result of charge flipping, i.e. the sign of the density below a threshold is reversed. Top curves: electron density after convergence (blue) and its flipped counterpart (red). Only the three atomic regions correspond to the true structure, small oscillations do not. See Oszlányi & Süto [Acta Cryst. (2004), A60, 134-141; (2005), A61, 147-152].
research papers





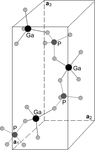
Electric-field-induced pseudoatomic displacements in α-GaPO4 are studied by means of an X-ray diffraction experiment. The site-selective analysis shows that the PO4 tetrahedra are more strongly distorted compared to those of GaO4 owing to the higher pseudoatomic charge of phosphorus.
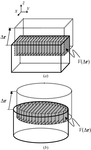
A development is presented that allows the simulation of reciprocal-space maps of epitaxic thin films exhibiting fluctuations in the size and shape of the crystalline domains over which diffraction is coherent. In all cases considered, the computation of the reciprocal-space map only requires a two-dimensional Fourier integral and the integrand has a simple analytical expression.
Open  access
access
access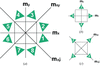
The orientational variants generated by twinning, precipitation or phase transition are algebraically described by a groupoid theory.
international union of crystallography
Free