journal menu
journal menu




issue contents
June 2008 issue
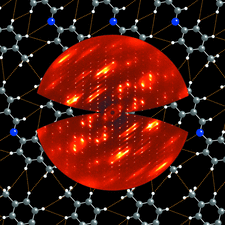
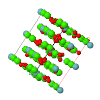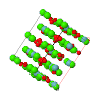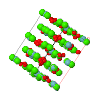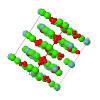
Cover illustration: The cover picture shows the strong and highly structured diffuse scattering from polymorph (II) of N-(p-methylbenzylidene)-p-methylaniline (MeMe) and indicates strongly correlated molecular motion not apparent from the Bragg analysis. Unlike polymorphs (I) and (III), which are disordered, polymorph (II) appears from Bragg scattering to be a perfectly normal ordered molecular crystal. The section of diffuse scattering data shown is part of a full three-dimensional data set recorded on beamline 1-ID-C at the Advanced Photon Source at Argonne, Illinois. Courtesy of Professor T. R. Welberry and Andrew Beasley.
research papers



Download citation





Download citation
The variation of structural and distortional parameters in synthetic olivine-type germanate compounds is studied, and a comparison is made with silicate olivine.
Download citation
Download citation
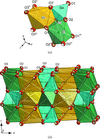
Vacancy ordering in AlB2-type ErGe2 − δ is described within a unified superspace approach. The observed powder diffraction data are indexed in the monoclinic superspace group X2m(α0γ)0s, X representing the centring vector (½,½,0,½). The q vector is shown to be a direct measure of the germanium occupancy as ErGe2 − α, with α in the range 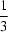 to ½.
to ½.
Download citation
Download citation
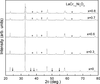
The crystal structure of perovskite LaCr1 − xNixO3 (0 ≤ x ≤ 1.0) at room temperature has been investigated by means of Rietveld refinement of X-ray diffraction data.

The coherence scattering lengths of a KY(WO4)2 single crystal in the directions parallel and perpendicular to the growth directions were estimated from the broadening of X-ray reflections. Qualitative analysis of the shape of the diffracted peaks and the intensity distribution in reciprocal space maps indicated the presence of dislocations and volume defects, which introduced a negative dilation into the crystal host.
Download citation
Download citation
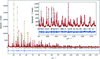
We report the crystal structure, electron density, electronic band dispersion and electronic density of states of samarium titanium oxysulfide (Sm2Ti2S2O4.9) photocatalyst obtained through the Rietveld analysis, maximum-entropy method (MEM) and MEM-based pattern fitting of the high-resolution synchrotron powder diffraction data taken at 298.7 K, in combination with the density-functional theory calculations.

The variation of the bond-valence sums (BVS) of the lanthanide atoms with the element number ZLn in isostructural garnet-type compounds Ln3Te2Li3O12 is strikingly similar to the variation of the third ionization energy of Ln with ZLn. This means that BVS does not represent the integer-number stoichiometric valence, but a non-integer structure-dependent property for which the term structural valence is proposed.
Download citation
Download citation
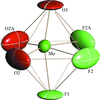
In highly dynamically disordered structures of (NH4)3MoO3F3 and (NH4)3WO3F3, it is possible to identify O and F atoms on a local scale using X-ray diffraction techniques. The true geometry of the distorted [MoO3F3]3− octahedron, which is rather far from C3v, explains the character of the observed vibrational spectra of the compounds.
Download citation
Download citation

The low-temperature phase III structures of perdeuterogermane and -stannane have been determined by high-resolution neutron powder diffraction at 5 K. Germane has an orthorhombic structure comprising molecules with C1 site symmetry and the structure of stannane is monoclinic with molecules located on twofold rotation axes.
Download citation
Download citation
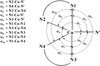
The four new distortion isomers of the five-coordinate [Cu(chelate)2X]+ cation have been compared by scatterplot analysis with 25 [Cu(chelate)2X]Y complexes of known crystal structure. Three of the present complexes are found to lie on an extended structural pathway towards the square-based pyramid.
Download citation
Download citation
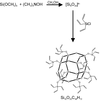
Crystal structures of two structural variants of octakis(trivinylsilyl)octasilicate, Si16O20C48H72, are reported. The paper illustrates both pseudo-symmetry and how the structure of a well ordered molecular compound can be used to determine the average structure of its disordered variant.
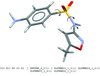
The hydrogen-bonding interactions of galanthamine, an anti-Alzheimer agent, have been analysed experimentally through observations in the Cambridge Structural Database and the Protein Data Bank and theoretically from ab initio and density functional theory calculations. Galanthamine interacts to a large degree through (i) multicentered hydrogen bonds involving its O atoms and (ii) polarized CH groups appearing as significant hydrogen-bond donors.
A method is described for the automated conformational clustering of complete molecules that occur in different crystal environments in the Cambridge Structural Database, by identifying all occurrences of a specific molecule either as the sole crystal component or co-crystallized with other component molecules or ions. The overall results of the methodology are tabulated and analysed in terms of molecular conformational diversity.
Download citation
Download citation
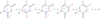
From a low-temperature (T = 20 K) high-resolution X-ray data set (sin θ/λ = 1.1 Å−1) the electron-density distribution of thymidine was derived by a classical multipole approach and by two invariom applications which considered the nearest and next-nearest neighbors for the heteroaromatic thymine ring. An electron localization function application yielded electron populations in the disynaptic valence basins which could be related to bond orders.
Download citation
Download citation

Variations in crystal and molecular structures, brought about by an intramolecular photochemical reaction of bi(anthracene-9,10-dimethylene), are described and explained.
Download citation
Download citation
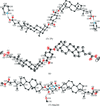
X-ray studies of three steroidal triads, A—B—A, reveal their wavy shape, inclusion properties and close resemblance to the crystal structures of cholic acids, and point to the special role of carboxylic acid/pyridine strong hydrogen bonds, and pyridine stacking interactions in the construction of one-dimensional polymeric tapes. The lithocholic acid triad:pyridine H:G crystals undergo a symmetry-lowering phase transition below 180 K.
short communications
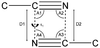
Database analysis shows that cyano groups form dipolar interactions in crystal structures, with the antiparallel dimer as the dominant motif. Ab initio calculations show this motif to have an attractive interaction energy similar to that of a medium-strength hydrogen bond.
Download citation
Download citation
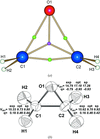
Weak intermolecular C—H⋯O interactions influence the electron density of ethylene oxide. This is visible on the Hirshfeld surface and in the polarization of the electrostatic potential.
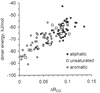
In organic carboxylic acid crystals the single and double carbon–oxygen bond lengths are variable and tend to be almost equal in aromatic acids. Reasons are tentatively discussed.