journal menu
journal menu




issue contents
June 2010 issue
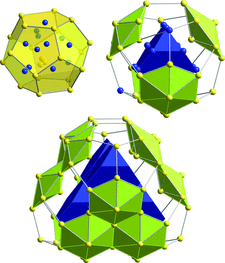
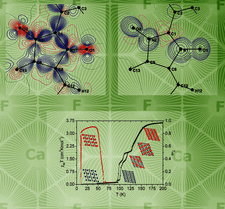
Cover illustration: The clusters shown on the cover image belong to the fundamental structural building units of cF(5928-x)-Al56.6Cu3.9Ta39.5, x = 20, one representative of a family of cluster-based tantalum copper aluminides with giant unit cells hosting up to more than 20 000 atoms. Their structures can be geometrically described as packings of clusters such as fullerenes, dodecahedra, pentagonal bifrusta, hexagonal bipyramids and Friauf polyhedra [Conrad et al. (2009). Acta Cryst. B65, 318-325; see also Weber et al. (2009). Acta Cryst. B65, 308-317].
research papers



Download citation





Download citation

An average statistical structure is presented for pyrrhotite 5C. An order–disorder structure description is proposed for the intermediate pyrrhotites of which 5C is an end member.
Download citation
Download citation

Anti-phase octahedral tilts are shown to be an essential structural feature of the high-pressure phase of both stoichiometric and heavily Ba-doped PbSc0.5Ta0.5O3 relaxor ferroelectrics with the perovskite structure type.
Download citation
Download citation

X-ray atomic orbital analysis of 4f and 5d electron configuration of SmB6 at 100, 165, 230 and 298 K
Electron density in SmB6 single crystals was measured at 100, 165, 230 and 298 K. The electron configuration of 2p of B, and 4f and 5d of Sm analysed by X-ray atomic orbital (XAO) explains well the observed electron density.
The improved values of the bond-valence parameters for the Sb3+/O2− ion pair, r0 = 1.927 Å and b = 0.446 Å, have been deduced from the crystal structures of the α and β polymorphs of Sb2O3 and from the set of precisely determined complex structures containing [Sb3+On] coordination shells.

The symmetry analysis of extinction rules in diffuse scattering is extended and applied to structure-dependent scattering data associated with `disordered' materials.
Download citation
Download citation
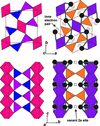
The structural compression of Bi2Ga4O9 was determined between 0 and 50 GPa across the high-pressure phase transition at 16 GPa from single-crystal X-ray diffraction and quantum mechanical calculations. The stereochemical activity of the lone electron pair of Bi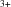 persists up to the highest pressure.
persists up to the highest pressure.
Open  access
access
access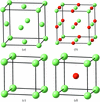
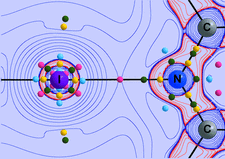
Theoretical calculations of the electron-localization function show that, at the volumes of the two CaO phases (rocksalt and CsCl type), the parent Ca structures (fcc: face-centred cubic; sc: simple cubic) exhibit charge-concentration zones which coincide with the positions occupied by the O atoms in their oxides. For the first time, the structure type, dimension and topology of CaO and BaSnO3 are explained in univocal physical terms.
Download citation
Download citation
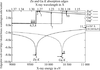
Synchrotron X-ray data collection at 11 wavelengths has allowed precise estimation of the zinc partial substitution at particular gallium sites in the gallophosphate ZnULM-5. A thorough exploration and comparison of available methodology is given, including the use of the CCP4 program suite, SHELXL and JANA2006. The heteroatom substitution is probably due to specific site relaxation.
CCDC reference: 782725
Download citation
Download citation
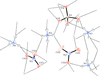
The crystal structures of two double salts of ammonium nitrate and ammonium sulfate are reported, as well as their application in the Rietveld quantification of nitrosulfate fertilizers.
Download citation
Download citation
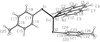
The experimental and theoretical charge densities in a photochromic light-emitting Zn complex indicate charge transfer to the phenanthroline ligand can originate from either thiolate ligand. The proper treatment of the Zn atom in charge-density analysis is described in detail.
CCDC reference: 782726
Download citation
Download citation
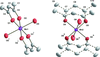
Four polymorphic forms of [Mn(acac)2(H2O)2]NO3·H2O are described. Lattice-energy calculations have been carried out to compare the stabilities of the polymorphs.
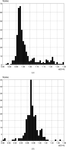
Mean X—H bond lengths for organic X = C, N, O, B and for a variety of terminal and homometallic bridging transition-metal hydrides are derived from the increased volume of neutron diffraction data now available in the Cambridge Structural Database. Revised default normalization distances for use in deformation density and hydrogen-bond studies are proposed for C—H, N—H and O—H bonds.
Download citation
Download citation
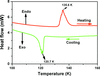
The reversible phase transition induced by temperature in pyridinium-3-carboxylic acid perchlorate was detected by differential scanning calorimetry, dielectric measurement and variable-temperature X-ray single-crystal diffraction analysis.
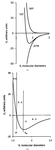
Intermolecular energies in organic crystals are classified with respect to strength and attractive or repulsive character. Energies provide a much better landscape than just intermolecular distances.