journal menu
journal menu




issue contents
December 2015 issue
Special issue on Energy Materials
Guest Editors: Karena W. Chapman, Simon Parsons and Richard I. Walton

Cover illustration: The packing arrangement of the zeolitic imidazolate framework ZIF-8 under ambient pressure conditions. ZnN4 tetrahedra are represented as solid green polyhedra. Colour key: C, black; H, grey [see McKellar & Moggach (2015). Acta Cryst. B71, 587-607]. We would like to thank both Mr Jorge Sotelo and Dr Alexander J. Graham for help in constructing the illustration.
editorial



Free 
Papers in this special issue focus on a broad range of research devoted to Energy Materials, investigating the role and application of structural science in one of the most important scientific challenges of our age: the identification of secure, sustainable and affordable sources of energy.
scientific commentaries
Free

Metal–organic frameworks (MOFs) demonstrate a wide variety of behavior in their response to pressure, including anomalous mechanical properties, negative linear compressibility, pressure-induced crystal-to-crystal and crystal-to-amorphous structural transitions. The discovery of framework materials combining novel pressure responses and high mechanical stability is key in the quest for applications of MOFs at the industrial level.
feature articles
Free
Here we present a review on the effect of high pressure on metal–organic framework materials.
energy materials
Open access
access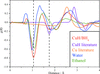
We show by a combination of diffraction and spectroscopic methods that CuH produced by borohydride reduction of a CuII salt consists of a core of CuH with the Wurtzite structure and a shell of water. We also demonstrate that the shell is exchangeable for ethanol.

Download citation




Download citation
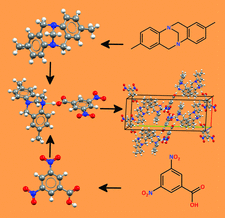
This paper reports on metal–organic frameworks which can be designed and synthesized to investigate structure predictability using a SBB (supermolecular building blocks) approach.
Single- and poly-cation inorganic homoleptic metal borohydrides are discussed on the basis of their structural relationship to metal oxides.
Open access
access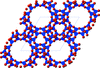
Framework materials possess intrinsic flexibility which can be investigated using geometric simulation. We review framework flexibility properties in energy materials and present novel results on the flexibility window of the EMT zeolite framework containing 18-crown-6 ether as a structure directing agent (SDA).

A combination of neutron powder diffraction and first-principles density functional theory calculations is used to understand the gas-sorption mechanisms of porous coordination polymers. This work overviews experimental and computational approaches taken to study these materials and the type of information that is obtained. This information includes the guest-responsive behaviour of the framework and the interaction between the host and the guest molecules, and between the guest molecules, that govern the gas sorption properties of the solid.
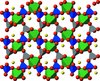
A carbon-coated lithium chromium polyanion, LiCrP2O7/C, was electrochemically tested versus lithium and found to be active with a specific charge of 105 mAh g−1 in the 1.5–4.5 V versus Li+/Li potential window. Operando X-ray diffraction experiments showed a reversible insertion reaction mechanism attributed to the LiCrIIIP2O7/Li2CrIIP2O7 couple leading to amorphization in the potential region of 1.8–2.2 V versus Li+/Li.
Download citation
Download citation
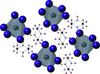
The structures of seven hybrid organic–inorganic lead iodide compounds and solvates, related to the photovoltaic material methylammonium lead iodide, have been elucidated. The dimensionality of the inorganic lead iodide component varies from zero-dimensional, discrete [PbI6]4− anions to two-dimensional [PbI4]2− sheets depending on the geometry and hydrogen-bonding behaviour of the counter organic molecular cation.
Download citation
Download citation
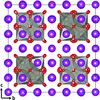
Type II Bi1 − xWxO1.5 + 1.5x is the first commensurate case among the known (3 + 3)-dimensional modulated δ-Bi2O3-related phases. The ability to refine a supercell model serves to validate the previously proposed crystal chemistry and modulated structure descriptions of these phases.
CCDC reference: 1429003
Download citation
Download citation

Here we address the use of operando diffraction techniques to understand the mechanisms involved for important electrode materials such as the spinels Li1 + xMn2 − xO4 (0 ≤ x ≤ 0.10) and LiNi0.4Mn1.6O4.
Open access
access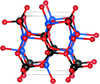
Structure–property relationships in Cu-based ternary oxides are explored using first-principles materials modelling.
Download citation
Download citation

The crystal structure of the higher manganese silicide MnSi1.7 is disordered incommensurate composite at temperatures not higher than 1273 K.
B-IncStrDB reference: 11402EFCaNO
CCDC reference: 1432489
Open access
access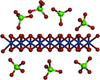
Amorphous thin-film oxygen evolving catalysts, OECs, of first-row transition metals show promise to serve as self-assembling materials in solar-driven, photoelectrochemical `artificial leaf' devices. This report demonstrates the ability to use high-energy X-ray scattering and atomic pair distribution function analyses, PDF, to resolve structure in amorphous metal oxide catalysts films, and is applied here to resolve domain structure differences induced by oxyanion substitution during the electrochemical assembly of amorphous cobalt oxide catalyst films.

An in-situ X-ray absorption spectroscopy comparison of nanostructured VO2 (B) positive intercalation electrodes synthesized by solvothermal and microwave assisted routes, highlighting the effect of morphological differences on electrochemical performance and oxidation state evolution, is presented.
Download citation
Download citation
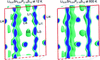
Based on neutron diffraction data, the present study reports the structural characteristics and related effects on conductivity for Li10GeP2S12 (LGPS)-type lithium superionic conductors.
scientific commentaries
Free

The development of a proper refinement algorithm that takes into account dynamical scattering guarantees, for electron crystallography, results approaching X-rays in terms of precision, accuracy and reliability. The combination of such dynamical refinement and electron diffraction tomography establishes a complete pathway for the structure characterization of single sub-micrometric crystals.
research papers

The method of structure refinement from electron diffraction tomography using dynamical diffraction theory is tested on a number of examples. General guidelines for the application of the method to new structures are provided.

The structurally most complex crystal structure of the sodium amalgam Na11Hg52 is best described via group-theoretic sublattices which are generated and concisely represented, including their point group symmetry, by number-theoretic formulae taken from the field of modular arithmetics developed by Gauß.
Download citation
Download citation
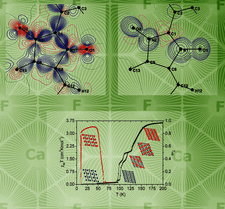
Hydrothermally grown single crystals of compositions ARE2F7 and ARE3F10 (A = K, Rb, Cs; RE = Y, La–Lu) have been studied by single-crystal X-ray diffraction. The study has identified a new structure type describing ARE2F7 compounds, and has extended the limits of the ARE3F10 structure types, while underscoring the importance of synthetic technique to directing the structure type.
Download citation
Download citation

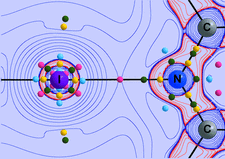
The similarities and differences between the electron density features in VB, V3B4 and VB2 derived from X-ray diffraction data as well as from theoretical calculations are analysed using quantum theory of atoms in molecules (QTAIM) descriptors.
Download citation
Download citation
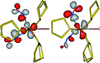
Photocrystallographic, IR analysis and DFT calculations reveal selective light-induced NO linkage isomerism in the dinitrosyl compound [RuBr(NO)2(PCyp3)2]BF4.
Download citation
Download citation
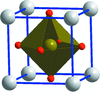
The crystal behavior of potassium bromate was studied up to 15 GPa pressure by both angle-dispersive X-ray diffraction experiments and ab initio total-energy calculations.
CCDC reference: 1427985
Download citation
Download citation
A temperature driven, single-crystal to single-crystal, first-order phase transition is reported and the structures and interaction schemes in both phases analyzed. A π⋯π ↔ C—H⋯π `switching' is deemed to be a possible driving force of the transition.