

 journal menu
journal menu





issue contents
February 2006 issue
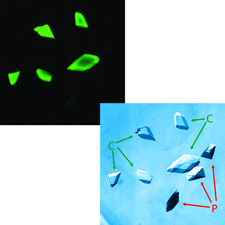
Cover illustration: Selective fluorescent staining of nucleic acid-containing crystals. A comparison of core Pol II protein crystals in the free form (P) and in complex with an 80-mer RNA inhibitor (C) is shown. On the right, a transmission light microscopic image reveals all crystals. On the left, a fluorescence image shows selective staining of nucleic acid complex crystals after incubation with SYBR-Gold (p. 146).
research papers






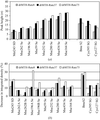
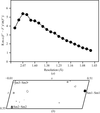
A systematic study of the specific radiation damage caused by different rates of X-ray irradiation is presented. The results suggest a small dose-rate related effect.
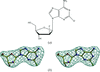
The crystal structure of human dCK in complex with clofarabine reveals the structural basis of activation by purine-nucleoside prodrugs.
PDB reference: dCK–clofarabine–ADP, 2a7q, r2a7qsf
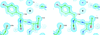
The crystal structure of T. gondii adenosine kinase in complex with an ATP analog at 1.1 Å resolution reveals that ATP binding and the formation of the induced anion hole can occur before adenosine binding.
PDB reference: AK–AMP-PCP, 2abs, 2abssf
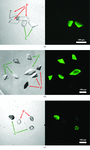
A highly sensitive and specific fluorescence-based assay for the rapid detection of nucleic acids in crystals of macromolecular complexes is described.

The structure of the manganese form of prostaglandin H2 synthase-1 reveals that metal substitution has no effect on the enzyme's structure; however, the manganese is displaced farther out of the porphyrin plane than is the native iron.
PDB reference: MnIII-reconstituted ovine PGHS-1, 2ayl, r2aylsf

The structure of the uropathogenic E. coli invasin DraD containing a C-terminal extension of 13 amino acids has been solved at 1.05 Å resolution. It forms dimers through the exchange of the C-terminal donor strands, resulting in a variant of the immunoglobulin fold.
PDB reference: DraD invasin, 2axw, r2axwsf
The X-ray crystal structure of the previously unknown bacteriophage P22 lysozyme was determined ab initio by direct methods using the program SIR2002. The asymmetric unit contained 2268 non-H protein atoms, making this one of the largest structures determined to date by ab initio direct methods.
Open  access
access
access
The structures of Dsk2 UBL, Dsk2 UBA and their complex are described. The complex explains the reduced affinity of the UBA domain for UBL compared with that for ubiquitin.
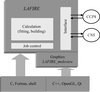
A manual intervention-free automatic refinement software package (LAFIRE) has been developed for high-throughput protein-structure analysis. This program begins from an initial model that can be approximate, fragmentary or even only main-chain and performs a whole refinement process to provide a final model including water molecules.

New crystal structures of glutathione transferase A1-1 show that the C-terminal region of the apo form can be ordered and helix-like. They also show how glutathione is bound in the absence of a second substrate and provide insight into the binding of a decarboxylated glutathione conjugate.
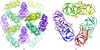
The highly pseudosymmetric crystal structure of HTLV-1 protease could be solved using low-sequence-identity models with Phaser but, with a single exception, not with standard application of other molecular-replacement programs.
PDB reference: HTLV-1 protease, 2b7f, r2b7fsf
short communications
Open access
access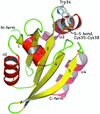
The high-resolution structure of thioredoxin from the parasite T. vaginalis has been determined. The redox-active disulfide has been partially photoreduced by the intense synchrotron radiation used in the analysis. Comparisons with human thioredoxin are presented.
PDB reference: thioredoxin, 2f51, r2f51sf
international union of crystallography
Free








