

 journal menu
journal menu





issue contents
February 2007 issue
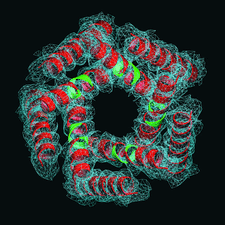
Cover illustration: 2Fo - Fc map (blue) computed from ab initio molecular replacement phases for the transmembrane region (red) of the mechanosensitive channel of large conductance (MscL) with omitted residues (green) (p. 188). The image was created with POVSCRIPT.
research papers




An in-house-developed automated imaging system for SBS microplate experiments has been designed and constructed, together with a web-based image-collection and storage database.



Substrate analogs induce an intermediate conformational change in Toxoplasma gondii adenosine kinase
T. gondii adenosine kinase substrate analogs with hydrophobic substituents at the 6-position result in a partially closed conformation of the enzyme.
PDB reference: adenosine kinase complex, 2a9y, r2a9ysf
The crystal structure of the cold-adapted catalase from the fish-pathogenic bacterium V. salmonicida (VSC) has been solved to 1.96 Å, and an extensive structural comparison of VSC and the catalase from the mesophilic human pathogen Proteus mirabilis has been performed.
PDB reference: catalase, 2isa, r2isasf

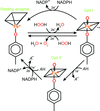
Crystals of aclacinomycin oxidoreductase are pseudomerohedrally twinned and are most likely to contain six twin domains. The structure was determined by multiwavelength anomalous diffraction using data from selenomethionine-substituted crystals.
PDB reference: aclacinomycin oxidoreductase, 2ipi, r2ipisf
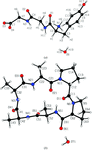
Application of the University at Buffalo theoretical databank of aspherical pseudoatoms to the refinement of experimental X-ray data from polypeptide structures significantly improves the resulting molecular geometries, residual Fourier difference maps, atomic anisotropic displacement parameters and phases of reflections. Implications for the refinement of protein data are discussed.
Open  access
access
access
A combined crystallographic, calorimetric and mutagenic study has been used to show how changes in pH give rise to two distinct binding modes of 2′-phospho-ADP-ribose to ketopantoate reductase.

A high-molecular-weight protease complex called trilobed protease (TLP) was recently discovered in the archaeon P. furiosus. The crystal structure of the N-terminal β-propeller domain of the trilobed protease at 2 Å resolution shows that the trilobed protease utilizes this accessory domain to control substrate access to the active site.
PDB reference: trilobed protease β-propeller domain, 2gop, r2gopsf
Open access
access
An ab initio molecular-replacement method for phasing X-ray diffraction data for symmetric helical membrane proteins has been developed. The described method is based on generating all possible orientations of idealized transmembrane helices and using each model in a molecular-replacement search.

The crystal structure of the putative mutarotase YeaD from S. typhimurium has been determined in orthorhombic and monoclinic crystal forms at 1.9 and 2.5 Å resolution, respectively. Comparison of the sequence and structure of YeaD with those of galactose mutarotases (GalMs) has allowed the identification of active-site residues and suggests plausible substrate specificity.
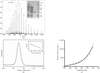
The crystal structure of a recombinant triosephosphate isomerase (TIM) from the archaeabacterium M. jannaschii has been determined at a resolution of 2.3 Å using X-ray diffraction data from a tetartohedrally twinned crystal. M. jannaschii TIM (MjTIM) is tetrameric, as is the case for two other structurally characterized archaeal TIMs, and the unliganded structure has a completely disordered active-site loop.
PDB reference: triosephosphate isomerase, 2h6r, r2h6rsf

The crystal structure of a binding-site mutant of NikA shows nickel binding to a second site.
PDB reference: mutant NikA, 2noo, r2noosf

Here, the crystal structure of Tt-dNTPase has been determined at 2.2 Å resolution, representing the first report of the tertiary structure of a dNTPase homologue belonging to the HD superfamily, a diverse group of metal-dependent phosphohydrolases that includes a variety of uncharacterized proteins.
PDB reference: dNTP triphosphohydrolase, 2dqb, r2dqbsf
Open access
access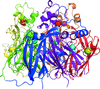
The three-dimensional molecular structure of human serum ceruloplasmin has been reinvestigated using X-ray synchrotron data collected at 100 K from a crystal frozen to liquid-nitrogen temperature.
PDB reference: ceruloplasmin, 2j5w, r2j5wsf

The crystal structure of M. tuberculosis caseinolytic protease subunit 1 (ClpP1) is reported. This structure shows novel inter-monomer arrangements that suggest a further source of flexibility for the inter-ring interactions of ClpP proteins.
short communications
Active p38α MAP kinase mutants caused severe difficulties during crystallization. The main hindrance was found to be protein heterogeneity, which was meticulously resolved by genetically modifying the recombinant protein and optimizing the expression and purification protocols. The success in obtaining crystallizable proteins strongly emphasizes that, in certain cases, high-throughput techniques together with low-throughput approaches, with careful monitoring and analysis of the results, are essential.
Open access
access
The structure of the DNA-binding domain of the response regulator VicR from E. faecalis has been solved at 1.9 Å resolution. It is very similar to the related domains from PhoB and OmpR, but differs in two loops that may affect transcription activation or DNA–protein interactions.
PDB reference: VicR DNA-binding domain, 2hwv, r2hwvsf

The issue of precision with respect to calculated solvent-accessible surface areas is addressed.







