

 journal menu
journal menu





issue contents
June 2007 issue
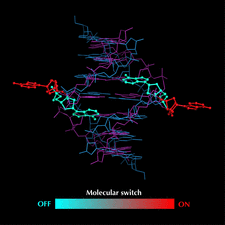
Cover illustration: DNA fragments of d(gcGAACgc) form a duplex, the adenosine residues bulging from which adopt two different conformations. One is folded back to interact with other part of the same duplex (blue) and the other is extended out to interact with the neighboring duplex (red), suggesting a molecular switch (p. 673).
research papers






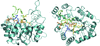
The crystal structure of human aldose reductase in complex with citrate at 0.82 Å resolution simultaneously shows the closed and open conformations of the safety belt. The two conformations correlate with different binding modes of the ligand.
PDB reference: aldose reductase–citrate complex, 2j8t, r2j8tsf
Two octamers with a sequence d(gcGAACgc) are associated to each other to form two types of bulge-containing duplexes different in conformation of the bulged-out adenosine residues, suggesting a molecular switch of the protruded adenosine residues.
PDB reference: d(gcGAACgc), 2got, r2gotsf

The X-ray structure of a parallel-stranded quadruplex has been solved to 1.5 Å resolution. The role of solvent molecules is analyzed.
PDB reference: 5′-(TGGGGT)-3′ quadruplex, 2o4f, r2o4fsf
Inhibition of the TF–FVIIa complex is seen as a promising target that is key to the development of clinical candidates for various cardiovascular applications. The crystal structure of the TF–FVIIa enzyme complex has been analyzed in order to design and synthesize small-molecule inhibitors.
PDB reference: TF–FVIIa–BCX-3607 complex, 2ec9, r2ec9sf
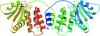
The structure of the dual-specificity H1 phosphatase encoded by the smallpox virus has been determined and used to identify small-molecule inhibitors by in silico screening methods.
PDB reference: Variola major H1 phosphatase, 2p4d, r2p4dsf

Molecular-dynamics simulations of a small protein allowed an experimentally verifiable optimization of TLS-group selection.
PDB reference: RM6 Rop deletion mutant, 1qx8, r1qx8sf

The crystal structure of the human complement protein CD59 is presented at 2.1 Å resolution. This structure is compared with a previous NMR model and the binding region for C8 and C9 is mapped.
PDB reference: CD59, 2ofs, r2ofssf

The structure of the phospholipase A2 toxin daboiatoxin from D. russelli siamensis (Myanmar) snake venom is reported.
PDB reference: daboiatoxin, 2h4c, r2h4csf
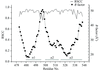
The 2.0 Å resolution crystal structure of hUBF HMG box5 has been solved using the single-wavelength anomalous-dispersion method, and the possible sites in hUBF HMG box5 which may interact with the first bromodomain in TAF1 were proposed.
PDB reference: hUBF HMG box 5, 2hdz, r2hzdsf
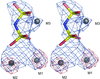
The structure of S. agalactiae family II inorganic pyrophosphatase in the presence of the competitive inhibitor imidodiphosphate was solved at 2.80 Å resolution by molecular replacement. Its structure is compared with those of other family II pyrophosphatases and structurally possible phosphorylation sites are discussed.
PDB reference: family II inorganic pyrophosphatase, 2enx, r2enxsf
short communications
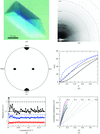
Pseudo-merohedral twinning in monoclinic crystals of human orotidine-5′-monophosphate decarboxylase
The crystal structure of orotidine-5′-monophosphate decarboxylase, the C-terminal domain of human UMP synthase, was determined by molecular replacement using data from highly pseudo-merohedrally twinned monoclinic crystals with a ≃ c unit-cell parameters.
book reviews
Free 








