journal menu
journal menu





issue contents
October 2009 issue
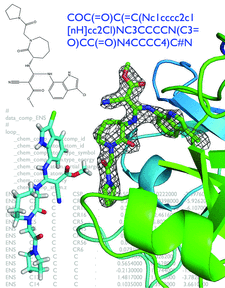
Cover illustration: Illustrations of steps in ligand handling as a part of a crystallographic structure determination (p. 1074). A SMILES string in the top right defines the chemistry of the ligand. The same ligand is shown by the two-dimensional representation in the top left. Below this is an optimized three-dimensional structure over a background of the restraints file in CIF format based on semi-empirical quantum chemistry calculations. To the right is the refined ligand structure in omit density. The protein structure (human Factor Xa) and data were obtained from the Protein Data Bank, entry 3ens. eLBOW was used to generate the restraints for the FXa inhibitor (ENS). The original refinement is explained in Shi et al.(2008) J. Med. Chem. 51, 7541-7551.
research papers








 access
access access
access access
access