

 journal menu
journal menu





issue contents
September 2011 issue
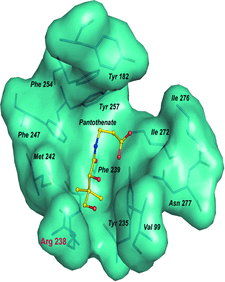
Cover illustration: van der Waals surface representation of the pantothenate binding site in the ATP initiation complex of M. tuberculosis pantothenate kinase (p. 774). The bound ligand is in the ball and stick representation.
research papers







New 96-well plates for crystallization have been developed that allow the collection of X-ray diffraction data directly in-plate or in situ. Automatic and rapid analyses of the data allow the detection of ligand bound to the crystallized macromolecule.

The crystal structure of a third form of bovine liver catalase, previously characterized by electron microscopy, has been determined using single-crystal X-ray diffraction. In light of this higher resolution data set, an assessment has been made of results from earlier electron-microscopy studies.
PDB reference: bovine liver catalase, 3nwl

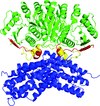
The crystal structures of human pyruvate dehydrogenase kinase 4 (PDK4) bound to the inhibitor M77976, as well as bound to AMPPNP or ADP, are presented. The structures provide a basis for the development of novel inhibitors targeting the nucleotide-binding pocket of PDK4.

The natural locations and conformations of pantothenate, pantothenol and N-nonylpantothenamide in M. tuberculosis pantothenate kinase derived from crystal structures provide further insight into the mode of action of this interesting enzyme.
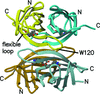
This study presents the crystal structure of the trifunctional THI20 from S. cerevisiae, an enzyme with an N-terminal HMP kinase (ThiD-like) domain and a C-terminal thiaminase II (TenA-like) domain. In addition, two distinct structural classes of TenA have been identified by comparison to other known structures.
PDB reference: THI20, 3rm5

Between T = 180 and 240 K, radiation damage progresses on minute timescales when the X-rays are off, suggesting that a fraction of damage at higher temperatures may be outrun using currently available sources and detectors.

An approach for ab initio phasing with de novo models has been developed by initiating molecular replacement during the course of the prediction of each model. This has reduced the computing time by about two orders of magnitude compared with performing molecular replacement after the generation of all models.
Analysis of atomic resolution crystal structures of wild-type streptavidin (1.03 Å) and its biotin complex (0.95 Å) indicate the range of conformational states taken on by this protein in the solid state. Most of the structural variation is found in the polypeptide loops between the strands in this β-sandwich protein.
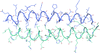
The crystal structure of the N-terminal fragment of the short nonmuscle α-tropomyosin has been determined at a resolution of 0.98 Å.
PDB reference: tropomyosin N-terminal fragment, 3azd
Open  access
access
access
The X-ray crystal structure (at 2.1 Å resolution) of an immunogen under development as part of a ricin vaccine for humans is presented and structure-based analysis of the results was conducted with respect to related proteins and the known determinants for inducing or suppressing the protective immune response.
PDB reference: RiVax, 3srp







