

 journal menu
journal menu





issue contents
October 2013 issue
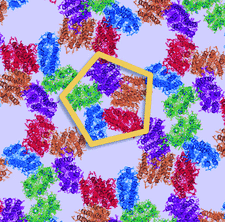
Cover illustration: The HOT75 D97N variant of blue-light-absorbing proteorhodopsin (p. 1965) forming pentameric rings in three-dimensional crystals composed of stacked two-dimensional crystals (type I membrane protein crystals).
scientific comment




The policy of the Protein Data Bank that the first deposition of a small-molecule ligand, even with erroneous atom numbering, sets a precedent over accepted nomenclature rules is disputed.
research papers


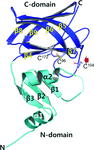
In this study, the crystal structure of CueP, a periplasmic copper-binding protein from Salmonella enterica, has been determined, revealing a V-shaped dimeric structure.
PDB reference: CueP, 4gqz

The crystal structures of two 1,4-dihydroxy-2-naphthoyl-CoA thioesterases of plant and cyanobacterial origin that are involved in the biosynthesis of phylloquinone are presented. The divergent structures of these two functionally similar enzymes indicate convergent evolution.
Open  access
access
access
The structure of the Tse3–Tsi3 complex associated with the bacterial type VI secretion system of P. aeruginosa has been solved and refined at 1.9 Å resolution. The structural basis of the recognition of the muramidase effector and its inactivation by its cognate immunity protein is revealed.
PDB reference: Tse3–Tsi3 complex, 3wa5

The diamagnetic levitation technique can aid in obtaining high-quality protein crystals. Comparison of the results obtained using three containerless conditions indicated that the diamagnetic levitation technique exhibited the greatest improvement in crystal quality, followed by that using agarose gel.
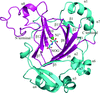
The crystal structure of JMJD5176-C revealed that three residues with large side chains located at the entrance to the substrate-binding pocket abolish the histone demethylase activity of this fragment of JMJD5. The structural similarity of JMJD5 to FIH suggested that human JMJD5 might function as a protein hydroxylase.
PDB reference: JMJD5, 4gaz

The notion of `resolution' is discussed and a formal definition applicable to various diffraction data sets, both complete and incomplete and both isotropic and anisotropic, is suggested.
Open access
access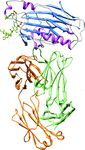
The gD–E317-Fab complex crystal revealed the conformational epitope of human mAb E317 on HSV gD, providing a molecular basis for understanding the viral neutralization mechanism.

The secreted success of cone-moulded weapons: unveiling the structural and functional details of the major collagenolytic and elastinolytic zinc-dependent proteinase from Aspergillus fumigatus.
PDB reference: A. fumigatus metalloprotease, 4k90
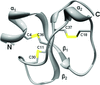
The crystal structure of the antitumoral and cell-penetrating polypeptide crotamine from C. durissus terrificus venom is reported.
PDB reference: crotamine, 4gv5
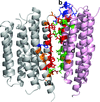
The first crystal structure of a proteorhodopsin shows a hexametric ring of protein in which the conserved photoactive-site histidine forms a hydrogen bond to the Schiff base proton acceptor from the same molecule and also to a tryptophan residue of a neighboring protomer. The structure and mutant studies reveal novel aspects of the ion translocation mechanism and cooperative behavior between protomers involving the inter-molecular His-Trp pair.
Open access
access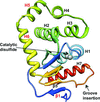
The gene product of M. tuberculosis Rv2969c is shown to be a disulfide oxidase enzyme that has a canonical DsbA-like fold with novel structural and functional characteristics.
PDB reference: Rv2969c, 4k6x

The first structures of DNA–protein complexes that include the DNA-binding domain from a LysR-type transcriptional regulator (LTTR) are described here and are used to explain the high degree of conservation in a DNA-sequence motif recognized by LTTRs (T-N11-A). Sequence-specific recognition depends primarily on direct protein–nucleotide base contacts.

The structure of α-phosphoglucomutase from L. lactis reveals overall structure similarity to eukaryotic phosphomannomutases, but with differences at the active site.
PDB reference: α-phosphoglucomutase, 4bnd
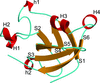
The first crystal structure of a barwin-like protein is determined at high resolution by single-wavelength anomalous diffraction phasing using the six intrinsic sulfur atoms present in the protein.
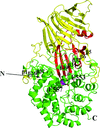
The crystal structure of the endo-β-1,3-glucanase from Rhizomucor miehei (RmLam81A) was determined in two crystal forms at 2.3 and 2.0 Å resolution, respectively. The structure of RmLam81A is greatly different from all endo-β-1,3-glucanase structures available.
Open access
access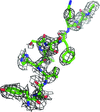
A genetic algorithm has been developed to optimize the phases of the strongest reflections in SIR/SAD data. This is shown to facilitate density modification and model building in several test cases.

A combination of small-angle X-ray scattering and atomic force microscopy reveals that the trimeric two-domain protein gephyrin adopts a multitude of compact and extended conformations, with a preference for compact states. This behaviour is caused by the long and primarily unstructured linker region, which also interacts with the structured domains located at either end of gephyrin. Potential functional implications of the structural data are discussed.
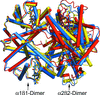
The crystallographic analysis of fully liganded Hb ζ2β2s trapped in a tense conformation is reported.
PDB reference: Hb ζ2β2s, 3w4u
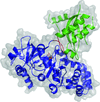
Combined small-angle X-ray scattering (SAXS) and crystallographic data were used to probe the two-step catalytic mechanism of the acyl acid–amido synthetase GH3.12. On-line size-exclusion chromatography directly upstream of the X-ray beam improved the sample homogeneity and allowed the determination of changes in the protein conformation in solution. X-ray models were fitted to the SAXS data to unambiguously determine the protein conformation in the presence of different ligands.
PDB reference: GH3.12–AMPCPP–salicylate complex, 4l39
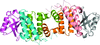
The structure of the complex between the N-terminal dimerization domain of Sgt2 and the ubiquitin-like domain of Get5 was determined in order to understand the interaction between Sgt2 and Get5 and to reveal how this complex is essential for the function of TA-protein delivery under stress conditions.
PDB reference: Sgt2N–Get5Ubl, 3zdm
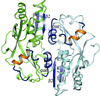
Differences in OxyR regulated expression of oxidative stress genes between Escherichia coli and Porphyromonas gingivalis are explained by very minor differences in structure and amino-acid sequence of the respective oxidized and reduced OxyR regulatory domains. These differences affect OxyR quaternary structures and are predicted from model building of full length OxyR–DNA complexes to confer distinct modes of DNA binding on this transcriptional regulator.
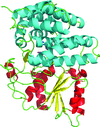
The crystal structure of the alcohol dehydrogenase (ADH) domain of an ADHE protein from G. thermoglucosidasius has been determined to 2.5 Å resolution. Docking of this with a homology model of the aldehyde dehydrogenase domain, for which no structure has been determined, has enabled the construction of a model of the complete ADHE and a putative mechanism of formation of the large multimeric `spirosomes' that have been reported for ADHE from this and other organisms.
PDB reference: ADH domain of ADHE, 3zdr
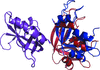
The intriguing crystal organization of the domain-swapped dimer of a human pancreatic ribonuclease variant indicates that the protein can form fibrils in solution. They were observed and characterized by atomic force microscopy. The importance of domain swapping in inducing native-like fibril formation is highlighted.
PDB reference: domain-swapped dimer of human pancreatic ribonuclease, 4kxh
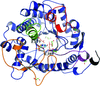
The structures of rice Os7BGlu26 β-D-mannosidase, of its product complex with β-D-mannose and of Os7BGlu26 computationally docked with 4-nitrophenyl β-D-mannoside suggest that the D-mannose pyranose ring makes a conformational transition from OS2 skew boat to B2,5 boat to 1S5 skew boat during the deglycosylation of the enzyme in hydrolysis. These structures led to the hypothesis that the residues interacting with the catalytic acid/base play a role in determining β-D-mannosidase activity, which was supported by site-directed mutagenesis and kinetic analysis.

Three crystal structures of the prolyl-tRNA synthetase domain of human glutamyl-prolyl-tRNA synthetase, two of which were complexed with small molecules, were determined by Zn-SAD phasing combined with molecular replacement. The structures revealed that conformational changes occur in the active site upon small-molecule binding.
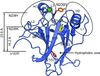
X-ray crystallographic structures of four p53 core-domain variants were determined in order to gain insights into the mechanisms by which certain second-site suppressor mutations rescue the function of a significant number of cancer mutations of the tumor suppressor protein p53.
short communications
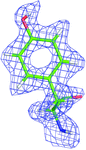
The first neutron protein structure refined against 1.75 Å resolution data collected on the new IMAGINE instrument at HFIR (ORNL) is reported.
PDB reference: perdeuterated rubredoxin, 4k9f
Different orientations of low-molecular-weight fragments in the binding pocket of a BRD4 bromodomain
Different orientations of similar low-molecular-weight fragments in the acetylated lysine-recognition pocket of the BRD4 N-terminal bromodomain are reported.







