

 journal menu
journal menu





issue contents
March 2015 issue
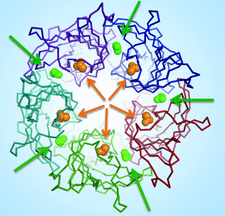
Cover illustration: Acetate binding sites of GLIC, a bacterial member of the pentameric Ligand-Gated Ion Channel (pLGIC) family (Fourati et al., p. 454). A top view of the receptor showing acetate molecules as either green (intersubunit acetate) or orange (intrasubunit acetate) spheres. Arrows show the entrance path to each site using the same colour scheme.
research papers






Open  access
access
access
The second crystal structure of a parasite protein preferentially enriched in the brain cyst of T. gondii has been solved at 2.75 Å resolution. Bradyzoite enolase 1 is reported to have differential functions as a glycolytic enzyme and a transcriptional regulator in bradyzoites.
PDB reference: Toxoplasma gondii enolase 1, 3otr
Open access
access
Crystallization of lysozyme with (R)-2-methyl-2,4-pentanediol produces more ordered crystals and a higher resolution protein structure than crystallization with (S)-2-methyl-2,4-pentanediol. The results suggest that chiral interactions with chiral additives are important in protein crystal formation.
The interaction between the A. fumigatus lectin AFL and biologically relevant oligosaccharides has been examined. The functional data were combined with detailed structures of the protein–saccharide complexes to understand the ligand binding at the atomic level.
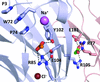
All acetate binding sites of GLIC, a bacterial member of the pentameric Ligand-Gated Ion Channel (pLGIC) family, have been mapped and its acetate-free structure determined. The two acetate binding sites overlap with known regulatory allosteric sites of other pLGICs.
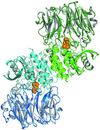
A mechanism for the dual function of oligopeptidases involves decoupling substrate binding and catalytic activity. Crystal structures of the A. pernix acylaminoacyl peptidase–covalent inhibitor complex provide insights into the role of substrates in this mechanism.

The crystal structure and functions of Hikeshi, a nuclear transport receptor of Hsp70s, are described.
Open access
access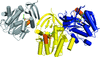
In this work, X-ray crystallography was used to examine ligand complexes of spermidine synthase from the malaria parasite Plasmodium falciparum (PfSpdS).
Open access
access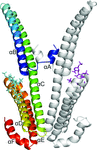
Two X-ray structures of APLP1 E2 with and without a heparin dodecasaccharide are presented, revealing two distinct binding modes of the protein to heparan sulfate. The data provide a mechanistic explanation of how APP-like proteins bind to heparan sulfates and how they specifically recognize nonreducing structures of heparan sulfates.
Two of the four conserved cysteine residues of an arsenic(III) S-adenosylmethionine methyltransferase bind aromatic arsenicals. The other two conserved cysteine residues form a disulfide bond.
Crystal structures of a GTP-bound SAMHD1 dimer and GTP/dNTP-bound SAMHD1 tetramers elucidate the mechanism of substrate-controlled allosteric regulation of SAMHD1 activated by GTP.
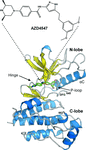
The complex between AZD4547 and the kinase domain of FGFR1 displays exquisite molecular recognition between ligand and protein, with the formation of an extended cavity by the closure of the P-loop.
PDB reference: FGFR1 in complex with AZD4547, 4wun
Open access
access
The first semi-liquid, non-protein nucleating agent for automated protein crystallization trials is described. This `smart material' is demonstrated to induce crystal growth and will provide a simple, cost-effective tool for scientists in academia and industry.
Open access
access
The tertiary structure of a β-lactamase derived from the halobacterium Chromohalobacter sp. 560 (HaBLA) was determined by X-ray crystallography. Three unique Sr2+-binding sites and one Cs+-binding site were discovered in the HaBLA molecule.
PDB references: β-lactamase, wild type, 3wrt; N288Q/N321Q mutant, 3wrz; 3ws0; 3ws1; 3ws2; 3ws4; 3ws5
Open access
access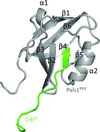
This study characterizes the interaction between the carboxy-terminal (ERLI) motif of the essential polarity protein Crb and the Pals1/Stardust PDZ-domain protein. Structures of human Pals1 PDZ with and without a Crb peptide are described, explaining the highly conserved nature of the ERLI motif and revealing a sterically blocked peptide-binding groove in the absence of ligand.
Open access
access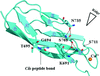
The surface properties and dynamics of PKD-like domains from ColG and ColH differ.
Open access
access
Four crystal structures of human LLT1, a ligand of human NKR-P1, are reported.
Open access
access
The crystal and solution structures of the T. thermophilus NlpC/P60 D,L-endopeptidase as well as the co-crystal structure of its N-terminal LysM domains bound to chitohexaose allow a proposal to be made regarding how the enzyme recognizes peptidoglycan.
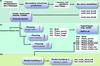
A fully automated procedure is presented that finds molecular-replacement solutions for coiled-coil structures based on models from de novo structure prediction.
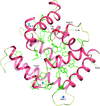
Thr-to-Asp mutations which mimic phosphorylation of the neuronal calcium-signalling protein calexcitin were introduced and the corresponding crystal structures were determined revealing that the C-terminal phosphorylation site (Thr188) exerts the greatest effects on the molecule's conformation. NMR and normal-mode dynamics suggest that the wild-type protein adopts a more open conformation in solution than crystallographic studies have previously indicated and further mutagenesis work indicated that the single calcium-binding site in the C-terminal domain of the protein is likely to have a sensory role.
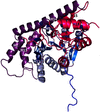
Two crystal structures of an aminotransferase from Psychrobacter sp. B6 have been determined at resolutions of 2.19 Å (native enzyme) and 2.76 Å (complex with aspartate). The protein was expressed in E. coli and specific activity tests indicated that aromatic amino acids and aspartate are the substrates.
Open access
access
The non-iterative feature-enhancing approach improves crystallographic maps' interpretability by reducing model bias and noise and strengthening the existing signal.
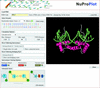
NuProPlot is a web-based automatic analysis and two-dimensional schematization program for protein–nucleic acid complexes. It can handle any type of nucleic acid, ranging from a few base pairs of simple DNA to thousands of RNA bases in complex protein–RNA structures, with user-friendly interactive options.
Open access
access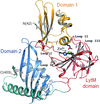
H. pylori Csd3 (HP0506), together with other peptidoglycan hydrolases, plays an important role in determining cell shape. Its crystal structure in the latent state is reported.

The crystal structure of apo ArnA features an unexpected central binding pocket and explains the strong cooperative effect observed in the dehydrogenase activity of ArnA.
PDB reference: full-length apo ArnA, 4wkg
Open access
access
A computer program that builds crystal structure models of nucleic acid molecules is presented. It can be accessed at http://iimcb.genesilico.pl/brickworx.

The growing profusion of spurious nonproline cis-peptide bonds in deposited structures suggests that a report on these should be added to the standard structural quality checks.
Open access
access
The structure of atrazine chlorohydrolase (AtzA) is presented and is used to reinterpret data from genetic, biochemical and evolutionary studies, providing insight into why this recently evolved enzyme appears to be poorly adapted for its physiological substrate compared with the alternative metal-dependent atrazine dechlorinase TrzN.
Open access
access
The crystal structures of farnesyl diphosphate synthase from the pathogen Pseudomonas aeruginosa in complex with substrates and inhibitors have been determined. The study reveals the presence of an allosteric binding pocket also in bacterial enzymes that, similar to the human enzyme, could be the target for the design of specific and potent inhibitors.
Open access
access
Co-crystal structures of P-glycoprotein with a series of engineered ligands reveal multiple ligand-binding modes, a ligand-binding site on the outer surface of the transporter and a conformational change that may couple to ATP hydrolysis.







