

 journal menu
journal menu




issue contents
July 2015 issue

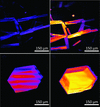
Cover illustration: C![[alpha]](/logos/entities/alpha_rmgif.gif) trace (cyan) of myoglobin (PDB entry 1a6g, essentially -helices) and the tube representation computed from the ScrewFrame parameters (Kneller & Hinsen, p. 1411).
trace (cyan) of myoglobin (PDB entry 1a6g, essentially -helices) and the tube representation computed from the ScrewFrame parameters (Kneller & Hinsen, p. 1411).
research papers




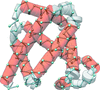
A generalization of the ScrewFit method for protein secondary-structure description and analysis is presented which uses only the positions of the Cα atoms and describes the winding of the protein main chain as a succession of screw motions which link consecutive residue-based Frenet frames on the discrete Cα space curve.

Download citation



Download citation
Open  access
access
access
The crystal structure of HMGB1 box A bound to an unmodified AT-rich DNA fragment is reported at a resolution of 2 Å. A new mode of DNA recognition for HMG box proteins is found in which two box A domains bind in an unusual configuration generating a highly kinked DNA structure.
PDB reference: HMGB box A bound to AT-rich DNA, 4qr9
The structure of the histidine kinase associated with the light–oxygen–voltage domain from B. abortus, containing 968 amino acids in the asymmetric unit, was solved using the `off-edge' anomalous signal from sulfur to a resolution of 2.90 Å by combining data from several isomorphous crystals (space group P21) as well as data collected from multiple orientations of the same crystal (true redundancy), stressing the importance of the latter approach in off-edge S-SAD. A second construct that has a shorter cloning artifact enabled the production of improved crystals and subsequent refinement of the structure to 2.51 Å resolution.
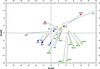
The correlation of histidine-ring geometry with the protonation state was analyzed in structures in the PDB and CSD. In conclusion, revised stereochemical restraints for histidine are proposed.
Download citation
Download citation
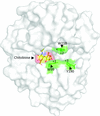
X-ray crystal structures of the apo, pseudo-apo form of human chitotriosidase (CHIT1) and its complex with chitobiose at atomic resolution combined with hybrid QM/MM calculations provide a detailed picture of CHIT1 hydrolysis mechanism. This novel mechanism involves changes in the conformation and protonation states of the conserved catalytic triad, as well as a new role for Tyr27 and conveys new insight onto the transglycosylation activity.
The intercalation of SHG-active dyes into formed protein crystals has allowed enhancement of the second-harmonic generation signal from protein crystals by ∼1000-fold.
Download citation
Download citation
Open access
access
The X-ray structure of protease-cleaved E. coli α-2-macroglobulin is described, which reveals a putative mechanism of activation and conformational change essential for protease inhibition.
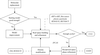
A direct-method-aided dual-space iterative phasing and model-building program suite, IPCAS, can be used to determine multi-subunit protein-complex structures starting from one or a partial subunit.
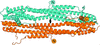
Controlled dehydration of IE1 crystals leads to molecular rearrangements that trigger a space-group transition from P21 to P43 with a concomitant reduction of the number of IE1 chains in the asymmetric unit. Analysis of the pre- and post-dehydration structures reveals a reshaping of the tertiary structure of IE, which possibly informs on the mechanism by which IE1 binds to host-cell target proteins.
Open access
access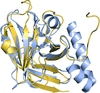
The structure of C. perfringens sortase D was determined at 1.99 Å resolution. Comparative biochemical and structural analyses revealed that this transpeptidase may represent a new subclass of the sortase D family.
PDB reference: C. perfringens sortase D, 4d70
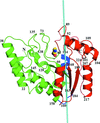
17 independent crystal structures of family I uracil-DNA glycosylase from M. tuberculosis (MtUng) and its complexes with uracil and its derivatives, distributed among five distinct crystal forms, provide information on conformational selection in DNA binding, segmental mobility and protein–ligand interactions.
PDB references: MtUng, form I, 4wpk; form IV, 4wrw; form V, 4wrx; complex with uracil, form I, 4wpl; form II, 4wru; form III, 4wrv; complex with 5-fluorouracil, form I, 4wry; 4wrz; form II, 4ws0; 4ws1; complex with 6-aminouracil, form I, 4ws2; form IV, 4ws3; complex with 5-nitrouracil, form I, 4ws4; form III, 4ws5; complex with 5-aminouracil, form I, 4ws6; complex with 5-chlorouracil, form II, 4ws7; complex with 2-thiouracil, form V, 4ws8

The crystal structure of the phosphatase domain of MTMR8 provides insights into its dimerization, membrane association and reversible oxidation.
PDB reference: MTMR8, 4y7i

Three crystal structures of L-galactitol-1-phosphate 5-dehydrogenase from E. coli have been solved. Functional data were combined with the structural results to understand the enantioselectivity of this enzyme.

The crystal structure of phosphatidylinositol 4-kinase (PI4K) IIβ reveals new details such as the high conformational heterogeneity of the lateral hydrophobic pocket and, together with the structure of PI4K IIα with a nucleoside analogue, provides a structural basis for isoform-specific inhibitor design.
Open access
access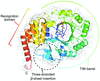
The crystal structure of a human dihydrouridine synthase, an enzyme associated with lung cancer, with 18% sequence identity to a T. maritima enzyme, has been determined at 1.9 Å resolution by molecular replacement after extensive molecular remodelling of the template.
PDB reference: human dihydrouridine synthase, 4xp7
Open access
access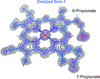
Crystal structures of the solubilized domain of cytochrome b5 from porcine liver were determined at sub-angstrom resolution in two crystal forms for both the oxidized and reduced states. The high-resolution structures provided information about the factors that are important for regulating the electronic properties of the haem group of cytochrome b5.







