journal menu
journal menu




issue contents
October 2020 issue

Cover illustration: Helical ensembles outperform ideal helices in molecular replacement. Examples of successful MR runs using members of a new library of helical ensembles. The solution of the transmembrane structure PDB entry 4ri2, with seven placed copies of the 25-residue helical ensemble (top); the globular structure PDB entry 5mq8, with 12 placed copies of the 15-residue helical ensemble (middle) and the solution for the coiled-coil structure PDB entry 1d7m, with two copies of the 35-residue ensemble (bottom). In all three images the red chains correspond to the deposited crystal structure and the blue chains to the MR-placed ensembles (only the first model of the ensemble is shown). Search models were made transparent to facilitate the visualization of the target structure. [Sánchez Rodríguez et al. (2020), Acta Cryst. D76, 962-970].
CCP4



 access
access access
access
 access
access
ISDSB2019



research papers

 access
access


 access
access
 access
access
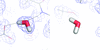 access
access