

 journal menu
journal menu





issue contents
April 2021 issue
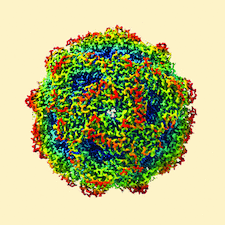
Cover illustration: A cryo-EM map, filtered and coloured by local resolution, produced by the Scipion plugin for the RELION software package [Sharov et al. (2021), Acta Cryst. D77, 403-410]. The plugin provides a set of Python wrappers for the RELION programs, while at the same time offering all of the capabilities of Scipion's image and metadata handling via a user-friendly graphical interface.
topical reviews




Free 
Knowledge of the structure and function of ion-channel proteins coded by coronaviruses lags behind the enormous progress on the other proteins of the SARS-CoV-2 virus, the etiological agent of COVID-19. The disease increasingly appears to constitute a complex syndrome of systemic breadth, making viral ion-channel proteins a possible target for therapeutic interventions.
CCP-EM

Open access
access
An overview of the Scipion plugin for the RELION software package is presented and various image-processing capabilities within the Scipion framework are discussed.
EMDB reference: reprocessed EMPIAR-10389 data (urease), EMD-12236

Open access
access
Sample preparation is a major limiting factor in cryo-electron microscopy (cryo-EM), particularly for small macromolecules under 100 kDa. Here, experiences in preparing kinesin-binding protein (KBP; 72 kDa) and KBP–kinesin motor domain (110 kDa) samples for cryo-EM are detailed, in particular some of the challenges and opportunities arising from surface interactions on the cryo-EM grid.
CCP4
Open access
access
Reparametrizing the positions and flexibility of protein models for refinement against diffraction data results in a substantial reduction in the number of parameters. This produces maps that are less affected by model bias, which allow the maps to more faithfully reflect the true crystal contents and reveal more biological information.
Open access
access
New forms of adaptive or top-out distance and torsion restraints are described that are suitable for restraining a model to match a reference structure during interactive rebuilding. In addition, their implementation in ISOLDE is described, along with some illustrative example applications.
Open access
access
Several approaches are presented to improve the performance of local correlation algorithms based on prior information about 3D search and target maps.
Open access
access
A procedure for the identification of a protein in a map from electron cryomicroscopy based on automated model building and sequence assignment is presented.
research papers
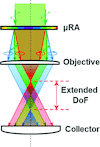
The addition of a single optical element enabled a 2.8-fold reduction of the data-acquisition time in crystal-screening processes based on optical imaging approaches.
Open access
access
This study reports the incorporation of small guest proteins by diffusion (soaking) into the solvent channels of a host protein crystal, visualized in time and space by microscopic techniques. The results represent a first step towards the realization of a protein-based crystal host (`hostal') system that may be used in the future as a carrier/entrainment system for protein guests and/or for guest-protein structure determination.

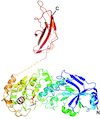
Crystal structures of human β-1,4-N-acetylglucosaminyltransferase 2, an enzyme essential for O-mannosylation, are reported, revealing a novel domain organization and providing the first rational basis for an explanation of the loss-of-function mutations observed in the clinic.
Open access
access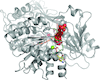
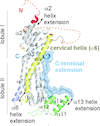
The crystal structure of Chaetomium thermophilum Prp2 bound to ADP-BeF3− and a poly-U12 RNA reveals a conserved pre-catalytic helicase conformation and only minor shifts of the C-terminal domains. Differences in the hook-loop and a loop of the helix-bundle domain compared with Prp43 evoke divergent conformations of the bound RNA, ultimately impacting the ability to unwind double-stranded RNA.
PDB reference: Prp2, complex with RNA and ADP-BeF3−, 6zm2
Biotin protein ligase from Leishmania major in complex with biotinyl-5′-AMP or biotin, crystallizes as a unique dimer, formed by cross-handshake interactions of its C-terminal domain.
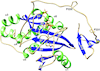
This study reveals the first crystal structure of a myosin XI globular tail domain and highlights the molecular adaptations that have allowed the evolution of specific mechanisms for the intracellular transport of vesicles and organelles in plant cells.
PDB reference: cargo-binding domain of class XI myosin, 7kfl
CRYO | EM
Open access
access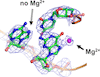
Cryo-EM can produce maps with resolutions that are sufficiently high that fine structural details can be discerned. Using calculated electrostatic potential maps, it is shown that it is possible to identify Mg2+ ions bound to nucleotide bases at which no charge compensation occurs, in contrast to Mg2+-bound phosphate groups, in the ribosomal RNA.
Open access
access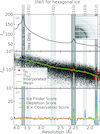
Analysis of diffraction data from three proteins indicates that the ice formed in internal crystal solvent is stacking-disordered. Application of a revised metric and algorithm for detecting ice from protein structure-factor data indicates that roughly 3.9% of PDB entries exhibit ice that is primarily hexagonal and 11.7% exhibit stacking-disordered ice.







