journal menu
journal menu




issue contents
June 2002 issue
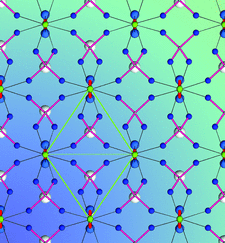
Cover illustration: The crystal structure of titanite, CaTiOSiO4, viewed down the direction of the octahedral TiO6 chains. The full green lines indicate a unit cell of the two-dimensional Ising model, used to predict diffuse scattering intensity observable in layers normal to the chain direction. Ti atoms are shown in green, Ca in light grey, O in blue. The thinner red lines are Si-O bonds, the thick red lines Ti-O1 bonds. Courtesy of T. Malcherek, C. Paulmann, M. C. Domeneghetti & U. Bismayer [J. Appl. Cryst. (2001), 34, 108-113].
research papers



The structure of KMgF3 has been determined by high-resolution neutron powder diffraction at 4.2 K, room temperature and at 10 K intervals from 373 K to 1223 K, and is found to remain cubic at all temperatures. Thermal expansion was fitted using a second-order Grüneisen approximation to the zero-pressure equation of state, while the atomic displacement parameters were found to increase smoothly with temperature and could be fitted using Debye models.

Download citation




Download citation
The crystal structure of the red polymorph of tetrahexylsexithiophene is solved from X-ray powder diffraction data, using a combination of direct-space Monte Carlo simulated annealing, DFT lattice-energy minimization and Rietveld refinement.
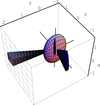
The application of the anisotropic Porod law is illustrated for the case of ellipsoidal particles.
Propagation of X-ray coherence through dynamical diffraction of a perfect crystal is discussed based on time-dependent Takagi–Taupin equations.

A microspectrophotometer is described that allows one to probe protein crystals both in absorption and in fluorescence mode. The results highlight the interest in fluorescence microspectrophotometry for future studies in structural enzymology and protein dynamics.
It is shown that the orientation distribution of the fibres in a composite can be determined by deconvolution of the crystallographic texture of the fibre phase in the composite with the crystallographic texture internal to the fibres.
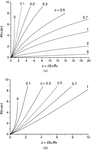
The size-broadened profile is modelled in terms of lognormal and gamma distributions of spherical crystallites. The method is applied to two ceria powders, which illustrates shortcomings of the current representation of the size-broadened profile in Rietveld refinement.
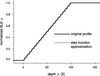
A novel free-form fit method for unbiased analysis of neutron and X-ray reflectivity data by an evolution strategy cancels out unsystematic features in scattering length density profiles, while it accentuates reliable details.
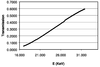
It is shown that incomplete absorption of the X-ray beam in the phosphor of an area detector causes an incident-angle dependence of the recorded X-ray intensities.
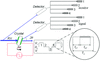
An instrument for applying strong electric fields to crystal samples during diffraction measurements is described.
short communications

Fractional coordinates for structures with known type can be modelled from first principles to about the accuracy of coordinates in routine powder diffraction studies. Powder patterns recalculated from published X-ray cell data and ab initio atom coordinates can then be used for identification of materials just as well as those recalculated from structure results coming from routine X-ray powder studies.
computer programs

A procedure is described that will match two or more sets of peaks or atoms related by different origins, enatiomorphs or symmetry elements.
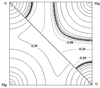
WinXPRO, working under the Windows operating system, enables the calculation of crystal and molecular properties using multipole parameters of the electron density.

The SnB interface provides users with several multiprocessing options for increasing throughput.
computer program abstracts

The general formalism for diffraction by crystals containing translational or rotational stacking faults is implemented in the program CALCIPOW.

A PC program that checks the symmetry properties of the unit cell and its contents has been added to the package Oscail.

The aim of the program is to generate a complete list of order parameters, both primary and secondary, associated with a given group-subgroup pair of space groups.
crystallographers
Free