 | Acta Crystallographica Section E Acta Crystallographica Section E CRYSTALLOGRAPHIC COMMUNICATIONS |

 journal menu
journal menu












research communications
| CRYSTALLOGRAPHIC COMMUNICATIONS |
 access
accessSynthesis, and thermal properties of dibromidobis(2-methylpyridine N-oxide-κO)cobalt(II)

aInstitut für Anorganische Chemie, Universität Kiel, Germany
*Correspondence e-mail: cnaether@ac.uni-kiel.de
Reaction of CoBr2 with 2-methylpyridine N-oxide in n-butanol leads to the formation of the title compound, [CoBr2(C6H7NO)2] or [CoBr2(2-methylpyridine N-oxide)2]. Its consists of one CoII cation as well as two bromide anions and two 2-methylpyridine N-oxide coligands in general positions. The CoII cations are tetrahedrally coordinated by two bromide anions and two 2-methylpyridine N-oxides, forming discrete complexes. In the these complexes are linked predominantly by weak C–H⋯Br hydrogen bonding into chains that propagate along the crystallographic a-axis. Powder X-ray diffraction (PXRD) measurements indicate that a pure phase was obtained. Thermoanalytical investigations prove that the title compound melts before decomposition; before melting, a further endothermic signal of unknown origin was observed that does not correspond to a phase transition.
Keywords: crystal structure; powder X-ray diffraction; synthesis; cobalt bromide; 2-methylpyridine N-oxide; differential thermoanalysis; differential scanning calorimetry.
CCDC reference: 2324135
1. Chemical context
Numerous transition-metal halide coordination compounds have been reported in the literature (Peng et al., 2010![]() ). Most of these compounds are characterized by metal halide substructures such as, for example, mono- and dinuclear units, chains or layers, that can be further linked by bridging ligands into 1-, 2- and 3-D networks (Peng et al., 2010
). Most of these compounds are characterized by metal halide substructures such as, for example, mono- and dinuclear units, chains or layers, that can be further linked by bridging ligands into 1-, 2- and 3-D networks (Peng et al., 2010![]() ; Näther et al., 2007
; Näther et al., 2007![]() ). We are especially interested in the thermal properties of such compounds because we have found that compounds with a high ratio between the metal halide and the ligands lose their ligands stepwise upon heating and transform into new compounds that usually show condensed metal–halide substructures (Näther et al., 2001
). We are especially interested in the thermal properties of such compounds because we have found that compounds with a high ratio between the metal halide and the ligands lose their ligands stepwise upon heating and transform into new compounds that usually show condensed metal–halide substructures (Näther et al., 2001![]() , 2002
, 2002![]() ; Näther & Jess, 2004
; Näther & Jess, 2004![]() ).
).
In this context, we have recently reported a new dinuclear complex with the composition [(CoBr2)2(2-methylpyridine N-oxide)4]·n-butanol in which the CoII cations are fivefold coordinated by two bromide anions and one terminal as well as two bridging 2-methylpyridine N-oxide ligands and linked into dinuclear units by two symmetry-related μ-1,1(O,O) 2-methylpyridine N-oxide coligands (Näther & Jess, 2023![]() ). The n-butanol solvate molecules can be removed by thermogravimetry, leading to the formation of a crystalline compound with the composition [CoBr2(2-methylpyridine N-oxide)2], for which the powder pattern is completely different from that of the pristine compound. We also found that the butanol molecules have already been lost upon storage at room-temperature, leading to the same crystalline phase as that obtained by thermal ligand removal. Moreover, the new crystalline phase shows two endothermic events before decomposition, which points to an interesting thermal behavior. Unfortunately, we were not able to solve its structure from PXRD data. Therefore, in the present work we performed a large number of crystallization experiments. The crystals obtained were characterized by single-crystal X-ray diffraction. The analysis proves that a new compound with the composition [CoBr2(2-methylpyridine N-oxide)2] was obtained, consisting of discrete complexes for which the calculated powder pattern is identical to that of the phase obtained by butanol removal from the dinuclear complex mentioned above. Larger amounts of a crystalline powder are easily available and comparison of the experimental powder pattern with that calculated from single crystal data proves that the title compound was obtained as a pure phase (Fig. 1
). The n-butanol solvate molecules can be removed by thermogravimetry, leading to the formation of a crystalline compound with the composition [CoBr2(2-methylpyridine N-oxide)2], for which the powder pattern is completely different from that of the pristine compound. We also found that the butanol molecules have already been lost upon storage at room-temperature, leading to the same crystalline phase as that obtained by thermal ligand removal. Moreover, the new crystalline phase shows two endothermic events before decomposition, which points to an interesting thermal behavior. Unfortunately, we were not able to solve its structure from PXRD data. Therefore, in the present work we performed a large number of crystallization experiments. The crystals obtained were characterized by single-crystal X-ray diffraction. The analysis proves that a new compound with the composition [CoBr2(2-methylpyridine N-oxide)2] was obtained, consisting of discrete complexes for which the calculated powder pattern is identical to that of the phase obtained by butanol removal from the dinuclear complex mentioned above. Larger amounts of a crystalline powder are easily available and comparison of the experimental powder pattern with that calculated from single crystal data proves that the title compound was obtained as a pure phase (Fig. 1![]() ), which allowed a detailed investigation of the thermal properties of the title compound to be undertaken.
), which allowed a detailed investigation of the thermal properties of the title compound to be undertaken.
![[Scheme 1]](yz2048scheme1.gif)
![[Figure 1]](yz2048fig1thm.gif) | Figure 1 Experimental (top) and calculated (bottom) powder patterns for the title compound. |
2. Structural commentary
The of the title compound, [CoBr2(2-methylpyridine N-oxide)2] (1), consists of one CoII cation, two bromide anions and two 2-methylpyridine N-oxide coligands that are located in general positions (Fig. 2![]() ). Compound 1 forms discrete complexes in which the CoII cations are fourfold coordinated by two bromide anions and two neutral 2-methylpyridine N-oxide coligands (Fig. 2
). Compound 1 forms discrete complexes in which the CoII cations are fourfold coordinated by two bromide anions and two neutral 2-methylpyridine N-oxide coligands (Fig. 2![]() ). Bond lengths and angles correspond to literature values and show that the tetrahedra are slightly distorted (Table 1
). Bond lengths and angles correspond to literature values and show that the tetrahedra are slightly distorted (Table 1![]() ). It is noted that the two known discrete tetrahedral complexes [CuCl2(2-methylpyridine N-oxide)2] and [ZnCl2(2-methylpyridine N-oxide)2] (refcodes QQQBVY and QQQBXY; Kidd et al., 1967
). It is noted that the two known discrete tetrahedral complexes [CuCl2(2-methylpyridine N-oxide)2] and [ZnCl2(2-methylpyridine N-oxide)2] (refcodes QQQBVY and QQQBXY; Kidd et al., 1967![]() ) are not isotypic to the title compound. For the latter compound, this is surprising because there are many examples in the literature where tetrahedral CoII and ZnII complexes are isotypic. On the other hand, there are very few examples reported in the literature where the thermodynamic relations between such complexes were fully investigated. It has been found, for example, that for two isotypic complexes the Co complex is thermodynamically stable at room temperature, whereas the isotypic Zn complex is metastable (Neumann et al., 2018
) are not isotypic to the title compound. For the latter compound, this is surprising because there are many examples in the literature where tetrahedral CoII and ZnII complexes are isotypic. On the other hand, there are very few examples reported in the literature where the thermodynamic relations between such complexes were fully investigated. It has been found, for example, that for two isotypic complexes the Co complex is thermodynamically stable at room temperature, whereas the isotypic Zn complex is metastable (Neumann et al., 2018![]() , 2019
, 2019![]() ).
).
| ||||||||||||||||||||||||||||||
![[Figure 2]](yz2048fig2thm.gif) | Figure 2 Crystal structure of the title compound with labeling and displacement ellipsoids drawn at the 50% probability level. |
3. Supramolecular features
In the of compound 1, a number of intermolecular C—H⋯O and C—H⋯Br contacts are observed, but most of the contacts show angles far from linearity, indicating that these correspond to very weak interactions (Table 2![]() ). However, a few of them exhibit distances and angles that point to intermolecular hydrogen bonding and, if they are considered as significant interactions, the discrete complexes are connected into chains propagating along the a-axis direction (Fig. 3
). However, a few of them exhibit distances and angles that point to intermolecular hydrogen bonding and, if they are considered as significant interactions, the discrete complexes are connected into chains propagating along the a-axis direction (Fig. 3![]() and Table 2
and Table 2![]() ).
).
|
| | Figure 3 Crystal structure of the title compound viewed along the a-axis. Intermolecular C—H⋯O and C—H⋯Br hydrogen bonds are shown as dashed lines. |
4. Thermoanalytical investigations
As mentioned above, recent investigations of the dinuclear complex tetrabromo-tetrakis(2-methylpyridine N-oxide)dicobalt(II) butanol solvate with thermogravimetry and differential thermoanalysis (TG-DTA) showed an endothermic signal after butanol removal where the sample mass did not change (Näther & Jess, 2023![]() ). Because it is the title complex that formed after solvent removal, its thermal properties were investigated in more detail using TG-DTA and DSC measurements (differential scanning calorimetry) as well as thermomicroscopy.
). Because it is the title complex that formed after solvent removal, its thermal properties were investigated in more detail using TG-DTA and DSC measurements (differential scanning calorimetry) as well as thermomicroscopy.
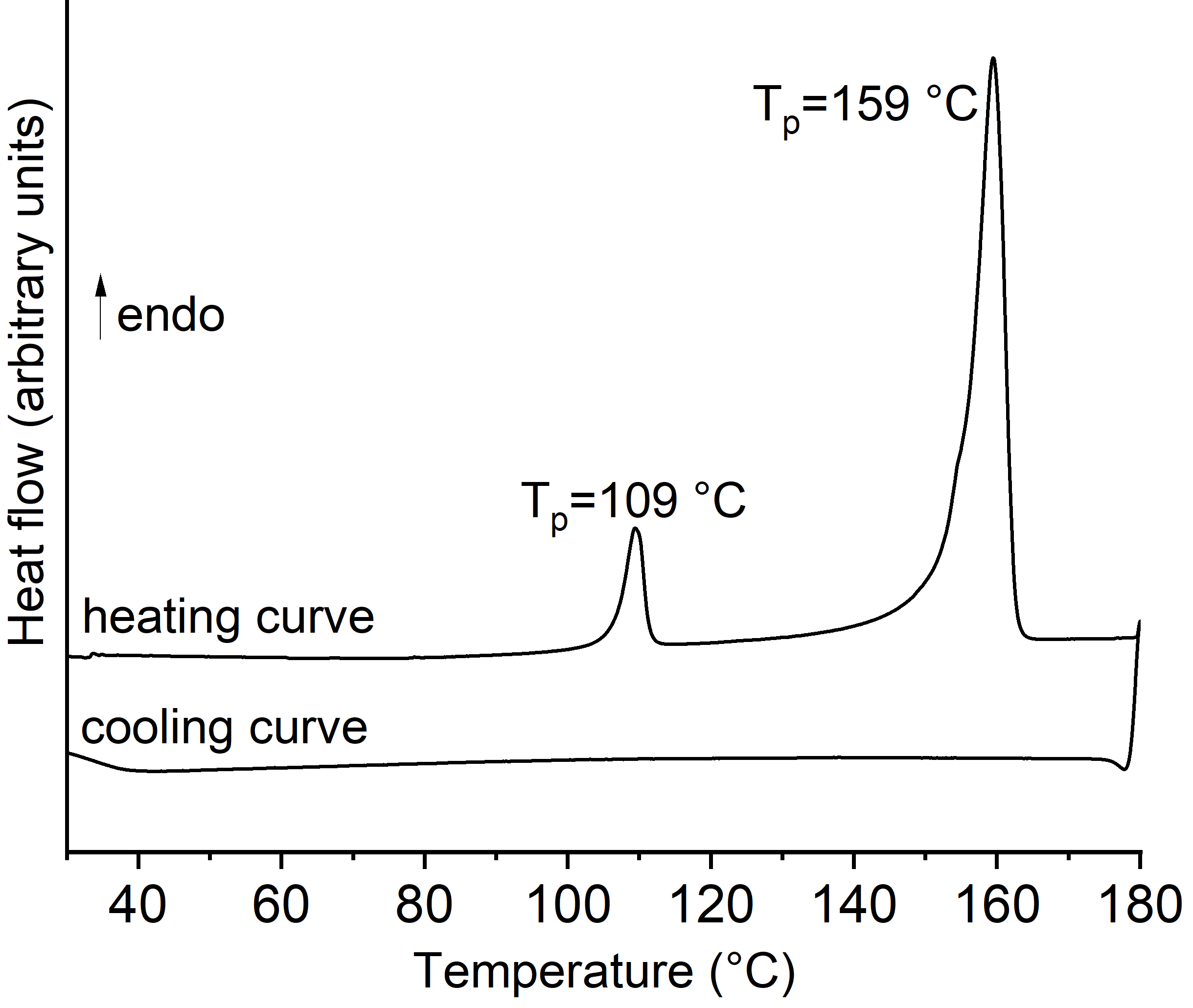
Upon heating, one poorly resolved mass loss is observed in the TG curve, which is accompanied by a strong exothermic event in the DTA curve at 278°C. The latter signal points to a decomposition of the 2-methylpyridine N-oxide ligand (Fig. S1). More importantly, before the first mass loss, two endothermic events at 109 and 155°C are observed in the DTA curve, which show that the overall thermal behavior is more complex. Therefore, DSC heating and cooling curves were measured, where two endothermic signals were observed upon heating (Fig. S2). Upon cooling, no exothermic signal was observed, which proves that the second endothermic event is irreversible. In contrast, if the title compound is measured up to 120°C and cooled down, an exothermic event is visible, which shows that this process is in principle reversible (Fig. 4![]() ). The same observations were made in the second heating and cooling run. However, the of these events continuously decreases, which means that this event is not entirely reversible.
). The same observations were made in the second heating and cooling run. However, the of these events continuously decreases, which means that this event is not entirely reversible.
![[Figure 4]](yz2048fig4thm.gif) | Figure 4 DSC heating and cooling runs for the title compound. |
The residues obtained at 120 and 180°C in the DSC measurements were investigated by powder X-ray diffraction (PXRD), which showed that the residue formed after the first endothermic event corresponds to the title complex (Fig. S3). No PXRD pattern could be measured for the residue formed after the second endothermic event because it adhered to the bottom of the crucible, indicative of melting. To investigate this in more detail, thermomicroscopic measurements were performed, which show melting at about 164°C (Fig. S5). This is in agreement with other tetrahedral Co but also Zn complexes, which melt upon heating (Neumann et al., 2018![]() , 2019
, 2019![]() ).
).
Finally, to investigate the origin of the first reversible endothermic event at 109°C, single-crystal measurements were performed between 23 and 167°C. Surprisingly, there are no structural changes and all data sets could be refined perfectly in P212121. The crystal decomposes upon further heating. The reason for this thermal event is therefore still unknown.
5. Database survey
A search of the CSD (version 5.43, last update March 2023; Groom et al., 2016![]() ) using CONQUEST (Bruno et al., 2002
) using CONQUEST (Bruno et al., 2002![]() ) reveals that no crystal structures of cobalt halide compounds with 2-methylpyridine N-oxide have been reported. As mentioned above, one compound with the composition [(CoBr2)2(2-methylpyridine N-oxide)4]·n-butanol was published recently (Näther & Jess, 2023
) reveals that no crystal structures of cobalt halide compounds with 2-methylpyridine N-oxide have been reported. As mentioned above, one compound with the composition [(CoBr2)2(2-methylpyridine N-oxide)4]·n-butanol was published recently (Näther & Jess, 2023![]() ) but does not yet appear as a hit.
) but does not yet appear as a hit.
For CuCl2 and ZnCl2, two discrete tetrahedral complexes with the composition [CuCl2(2-methylpyridine N-oxide)2] and [ZnCl2(2-methylpyridine N-oxide)2] have been reported, but neither of them is isotypic to the title compound (refcodes QQQBVY and QQQBXY; Kidd, et al., 1967![]() ). Similar complexes with a tetrahedral coordination are also reported with CuCl2 and ZnCl2 and 3-methylpyridine N-oxide and 4-methylpyridine, respectively, as ligands [QQQBWA, QQQBWA01, QQQBXM (Kidd et al., 1967
). Similar complexes with a tetrahedral coordination are also reported with CuCl2 and ZnCl2 and 3-methylpyridine N-oxide and 4-methylpyridine, respectively, as ligands [QQQBWA, QQQBWA01, QQQBXM (Kidd et al., 1967![]() ), CMPOCU (Watson & Johnson, 1971
), CMPOCU (Watson & Johnson, 1971![]() ), and CMPOCU01, QQQBXG (Kidd et al., 1967
), and CMPOCU01, QQQBXG (Kidd et al., 1967![]() )]. Finally, [ZnI2(4-methylpyridine N-oxide)2] also forms a tetrahedral complex (SANRUV; Shi et al., 2005
)]. Finally, [ZnI2(4-methylpyridine N-oxide)2] also forms a tetrahedral complex (SANRUV; Shi et al., 2005![]() ).
).
There are additional compounds with different structures and 2-methylpyridine N-oxide as ligand, including [(CuCl2)3(2-methylpyridine N-oxide)2(H2O)2] (PIOCUA; Sager & Watson, 1968![]() ), [MnCl2(2-methylpyridine N-oxide)(H2O)] (VEJMAB; Kang et al., 2017
), [MnCl2(2-methylpyridine N-oxide)(H2O)] (VEJMAB; Kang et al., 2017![]() ), and [(MnBr2)2(2-methylpyridine N-oxide)2(H2O)4] bis(2-methylpyridine N-oxide) solvate (VONHEO; Lynch et al., 2019
), and [(MnBr2)2(2-methylpyridine N-oxide)2(H2O)4] bis(2-methylpyridine N-oxide) solvate (VONHEO; Lynch et al., 2019![]() ).
).
Lastly, 2-methylpyridine N-oxide in its protonated cationic form together with a tetrachloro aurate(III) anion and a neutral 2-methylpyridine N-oxide (CICBIZ; Hussain & Aziz Al-Hamoud, 1984![]() ) and Co(2-methylpyridine N-oxide)5 with two ClO4− counter-ions [PICOCO (Coyle & Ibers, 1970
) and Co(2-methylpyridine N-oxide)5 with two ClO4− counter-ions [PICOCO (Coyle & Ibers, 1970![]() ) and PICOCO01 (Bertini et al., 1975
) and PICOCO01 (Bertini et al., 1975![]() )] have been reported.
)] have been reported.
6. Synthesis and crystallization
CoBr2 (97%) was purchased from Alfa Aesar and 2-methylpyridine N-oxide (98%) was obtained from Thermo Scientific.
Synthesis:
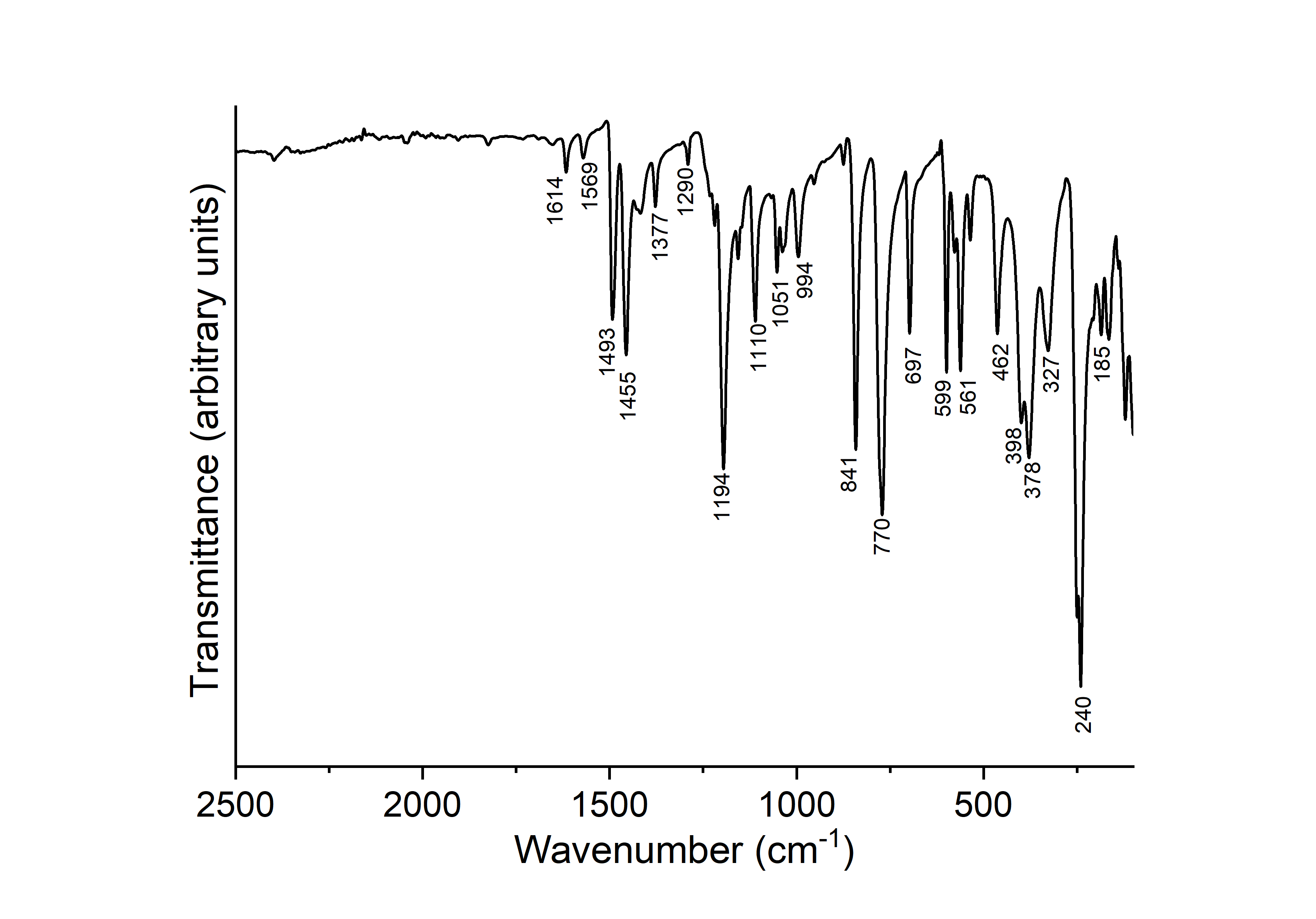
109 mg CoBr2 (0.5mmol) and 218 mg 2-picoline N-oxide (2 mmol) were stirred for 1 d in n-butanol at room temperature. The precipitate was filtered off and dried in air. Single crystals were obtained using the same conditions but without stirring. N.B. When stoichiometric amounts were used, in some batches a very small amount of the known compound [CoBr2]2(2-methylpyridine N-oxide)4·n-butanol was found (Näther & Jess, 2023![]() ). The IR spectrum of the title compound is shown in Fig. S5.
). The IR spectrum of the title compound is shown in Fig. S5.
Experimental details:
The PXRD measurements were performed with a Stoe Transmission Powder Diffraction System (STADI P) equipped with a MYTHEN 1K detector and a Johansson-type Ge(111) monochromator using Cu Kα1 radiation (λ = 1.540598 Å). Thermogravimetry and differential thermoanalysis (TG-DTA) measurements were performed in a dynamic nitrogen atmosphere in Al2O3 crucibles using a STA-PT 1000 thermobalance from Linseis. The instrument was calibrated using standard reference materials. measurements were performed with a DSC from Mettler Toledo in Al pans under nitrogen atmosphere with 10°C min−1. The IR spectra were measured using an ATI Mattson Genesis Series FTIR Spectrometer, control software: WINFIRST, from ATI Mattson.
7. Refinement
Crystal data, data collection and structure details are summarized in Table 3![]() . C-bound hydrogen atoms were positioned with idealized geometry (methyl H atoms allowed to rotate but not to tip) and were refined isotropically with Uĩso(H) = 1.2 Ueq(C) (1.5 for methyl hydrogen atoms) using a riding model. One reflection (outlier) was removed using the OMIT command.
. C-bound hydrogen atoms were positioned with idealized geometry (methyl H atoms allowed to rotate but not to tip) and were refined isotropically with Uĩso(H) = 1.2 Ueq(C) (1.5 for methyl hydrogen atoms) using a riding model. One reflection (outlier) was removed using the OMIT command.
|
Supporting information
CCDC reference: 2324135
contains datablock I. DOI: https://doi.org/10.1107/S2056989024000252/yz2048sup1.cif
Structure factors: contains datablock I. DOI: https://doi.org/10.1107/S2056989024000252/yz2048Isup2.hkl
DTG, TG and DTA curve for the title compound. DOI: https://doi.org/10.1107/S2056989024000252/yz2048sup3.png
DSC heating and cooling curve of the title compound measured to 180C with 10C/min. DOI: https://doi.org/10.1107/S2056989024000252/yz2048sup4.png
Experimental powder pattern of the residue obtained at 120C in a DSC measurement of the title compound. DOI: https://doi.org/10.1107/S2056989024000252/yz2048sup5.png
Microscopic images of the title compound at different temperatures. DOI: https://doi.org/10.1107/S2056989024000252/yz2048sup6.jpg
IR spectrum of the title compound. The values of the most prominent vibrations are given. DOI: https://doi.org/10.1107/S2056989024000252/yz2048sup7.png
| [CoBr2(C6H7NO)2] | Dx = 1.919 Mg m−3 |
| Mr = 437.00 | Cu Kα radiation, λ = 1.54184 Å |
| Orthorhombic, P212121 | Cell parameters from 7293 reflections |
| a = 7.6106 (2) Å | θ = 3.5–78.6° |
| b = 7.8024 (2) Å | µ = 15.09 mm−1 |
| c = 25.4699 (5) Å | T = 100 K |
| V = 1512.43 (6) Å3 | Needle, blue |
| Z = 4 | 0.18 × 0.04 × 0.03 mm |
| F(000) = 852 |
| XtaLAB Synergy, Dualflex, HyPix diffractometer | 3235 independent reflections |
| Radiation source: micro-focus sealed X-ray tube, PhotonJet (Cu) X-ray Source | 3183 reflections with I > 2σ(I) |
| Mirror monochromator | Rint = 0.030 |
| Detector resolution: 10.0000 pixels mm-1 | θmax = 80.5°, θmin = 3.5° |
| ω scans | h = −9→9 |
| Absorption correction: multi-scan (CrysAlisPro; Rigaku OD, 2022) | k = −9→7 |
| Tmin = 0.448, Tmax = 1.000 | l = −32→31 |
| 9313 measured reflections |
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.036 | w = 1/[σ2(Fo2) + (0.0482P)2 + 3.0792P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.093 | (Δ/σ)max = 0.001 |
| S = 1.06 | Δρmax = 0.65 e Å−3 |
| 3235 reflections | Δρmin = −0.47 e Å−3 |
| 174 parameters | Absolute structure: Flack x determined using 1248 quotients [(I+)-(I-)]/[(I+)+(I-)] (Parsons et al., 2013) |
| 0 restraints | Absolute structure parameter: −0.026 (5) |
| Primary atom site location: dual |
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| x | y | z | Uiso*/Ueq | ||
| Co1 | 0.42209 (13) | 0.36450 (12) | 0.40064 (4) | 0.0302 (2) | |
| Br1 | 0.73569 (8) | 0.36730 (8) | 0.39846 (2) | 0.03254 (16) | |
| Br2 | 0.29398 (9) | 0.59533 (9) | 0.35188 (3) | 0.04006 (18) | |
| O1 | 0.3404 (6) | 0.1401 (6) | 0.37462 (16) | 0.0350 (9) | |
| N1 | 0.2739 (7) | 0.1236 (7) | 0.32603 (18) | 0.0320 (10) | |
| C1 | 0.3749 (9) | 0.0537 (8) | 0.2878 (3) | 0.0337 (13) | |
| C2 | 0.3008 (10) | 0.0343 (9) | 0.2389 (3) | 0.0380 (14) | |
| H2 | 0.369838 | −0.011608 | 0.211185 | 0.046* | |
| C3 | 0.1283 (10) | 0.0798 (10) | 0.2291 (3) | 0.0401 (15) | |
| H3 | 0.078032 | 0.062728 | 0.195331 | 0.048* | |
| C4 | 0.0306 (10) | 0.1505 (10) | 0.2693 (3) | 0.0410 (15) | |
| H4 | −0.087882 | 0.183703 | 0.263439 | 0.049* | |
| C5 | 0.1060 (9) | 0.1723 (9) | 0.3177 (3) | 0.0375 (14) | |
| H5 | 0.039908 | 0.221735 | 0.345488 | 0.045* | |
| C6 | 0.5573 (10) | 0.0018 (9) | 0.3022 (3) | 0.0405 (15) | |
| H6A | 0.612177 | 0.092720 | 0.323040 | 0.061* | |
| H6B | 0.625929 | −0.017063 | 0.270125 | 0.061* | |
| H6C | 0.553525 | −0.104245 | 0.322723 | 0.061* | |
| O11 | 0.3208 (6) | 0.3399 (6) | 0.47074 (16) | 0.0345 (9) | |
| N11 | 0.3415 (9) | 0.4564 (7) | 0.5087 (2) | 0.0406 (14) | |
| C11 | 0.2092 (11) | 0.5484 (9) | 0.5266 (3) | 0.0414 (15) | |
| C12 | 0.2428 (12) | 0.6598 (8) | 0.5683 (3) | 0.0441 (17) | |
| H12 | 0.148608 | 0.726718 | 0.581688 | 0.053* | |
| C13 | 0.3997 (12) | 0.6768 (10) | 0.5901 (3) | 0.0497 (18) | |
| H13 | 0.417117 | 0.755090 | 0.618185 | 0.060* | |
| C14 | 0.5407 (11) | 0.5771 (10) | 0.5711 (3) | 0.0460 (17) | |
| H14 | 0.653938 | 0.586838 | 0.586508 | 0.055* | |
| C15 | 0.5133 (10) | 0.4686 (10) | 0.5312 (3) | 0.0406 (15) | |
| H15 | 0.606829 | 0.400540 | 0.518027 | 0.049* | |
| C16 | 0.0416 (10) | 0.5222 (10) | 0.5006 (3) | 0.0462 (17) | |
| H16A | 0.052604 | 0.552188 | 0.463370 | 0.069* | |
| H16B | −0.047766 | 0.595112 | 0.516994 | 0.069* | |
| H16C | 0.006869 | 0.401760 | 0.503838 | 0.069* |
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Co1 | 0.0325 (5) | 0.0338 (5) | 0.0241 (4) | −0.0023 (4) | 0.0002 (4) | −0.0012 (4) |
| Br1 | 0.0320 (3) | 0.0355 (3) | 0.0301 (3) | −0.0009 (2) | −0.0015 (2) | 0.0013 (2) |
| Br2 | 0.0382 (3) | 0.0437 (4) | 0.0383 (3) | 0.0048 (3) | −0.0011 (3) | 0.0066 (3) |
| O1 | 0.046 (2) | 0.035 (2) | 0.0242 (19) | −0.007 (2) | −0.0032 (17) | −0.0020 (17) |
| N1 | 0.036 (2) | 0.034 (2) | 0.026 (2) | −0.008 (2) | 0.0017 (19) | −0.0017 (19) |
| C1 | 0.035 (3) | 0.033 (3) | 0.033 (3) | −0.002 (2) | 0.004 (2) | 0.002 (2) |
| C2 | 0.045 (4) | 0.039 (3) | 0.030 (3) | −0.004 (3) | 0.002 (3) | −0.002 (2) |
| C3 | 0.046 (4) | 0.042 (4) | 0.033 (3) | −0.006 (3) | −0.005 (3) | −0.003 (3) |
| C4 | 0.037 (3) | 0.042 (4) | 0.044 (4) | 0.000 (3) | −0.003 (3) | −0.001 (3) |
| C5 | 0.037 (3) | 0.036 (3) | 0.039 (3) | 0.000 (3) | 0.007 (3) | −0.010 (3) |
| C6 | 0.040 (4) | 0.042 (4) | 0.039 (4) | 0.002 (3) | −0.002 (3) | −0.003 (3) |
| O11 | 0.043 (2) | 0.033 (2) | 0.0273 (19) | −0.0040 (19) | 0.0015 (18) | −0.0040 (17) |
| N11 | 0.064 (4) | 0.030 (3) | 0.028 (2) | −0.004 (3) | 0.010 (3) | 0.000 (2) |
| C11 | 0.048 (4) | 0.037 (3) | 0.039 (3) | −0.002 (3) | 0.001 (3) | 0.005 (3) |
| C12 | 0.065 (5) | 0.032 (3) | 0.035 (3) | −0.002 (3) | 0.001 (3) | −0.002 (2) |
| C13 | 0.056 (5) | 0.040 (4) | 0.053 (4) | 0.000 (3) | 0.008 (4) | −0.003 (3) |
| C14 | 0.049 (4) | 0.047 (4) | 0.042 (4) | −0.004 (3) | −0.006 (3) | 0.003 (3) |
| C15 | 0.045 (4) | 0.040 (3) | 0.037 (3) | 0.010 (3) | 0.007 (3) | 0.002 (3) |
| C16 | 0.044 (4) | 0.050 (4) | 0.045 (4) | −0.012 (3) | −0.004 (3) | 0.007 (3) |
| Co1—Br1 | 2.3874 (11) | C6—H6B | 0.9800 |
| Co1—Br2 | 2.3951 (11) | C6—H6C | 0.9800 |
| Co1—O1 | 1.973 (5) | O11—N11 | 1.336 (7) |
| Co1—O11 | 1.954 (4) | N11—C11 | 1.318 (10) |
| O1—N1 | 1.343 (6) | N11—C15 | 1.431 (10) |
| N1—C1 | 1.355 (8) | C11—C12 | 1.396 (10) |
| N1—C5 | 1.350 (9) | C11—C16 | 1.452 (11) |
| C1—C2 | 1.376 (10) | C12—H12 | 0.9500 |
| C1—C6 | 1.491 (10) | C12—C13 | 1.323 (12) |
| C2—H2 | 0.9500 | C13—H13 | 0.9500 |
| C2—C3 | 1.383 (10) | C13—C14 | 1.411 (12) |
| C3—H3 | 0.9500 | C14—H14 | 0.9500 |
| C3—C4 | 1.380 (10) | C14—C15 | 1.339 (11) |
| C4—H4 | 0.9500 | C15—H15 | 0.9500 |
| C4—C5 | 1.371 (10) | C16—H16A | 0.9800 |
| C5—H5 | 0.9500 | C16—H16B | 0.9800 |
| C6—H6A | 0.9800 | C16—H16C | 0.9800 |
| Br1—Co1—Br2 | 112.83 (4) | H6A—C6—H6B | 109.5 |
| O1—Co1—Br1 | 108.39 (15) | H6A—C6—H6C | 109.5 |
| O1—Co1—Br2 | 111.40 (14) | H6B—C6—H6C | 109.5 |
| O11—Co1—Br1 | 114.62 (14) | N11—O11—Co1 | 123.2 (4) |
| O11—Co1—Br2 | 112.76 (15) | O11—N11—C15 | 116.3 (6) |
| O11—Co1—O1 | 95.48 (18) | C11—N11—O11 | 122.1 (7) |
| N1—O1—Co1 | 120.9 (4) | C11—N11—C15 | 121.5 (6) |
| O1—N1—C1 | 119.1 (5) | N11—C11—C12 | 117.6 (7) |
| O1—N1—C5 | 118.3 (5) | N11—C11—C16 | 115.8 (7) |
| C5—N1—C1 | 122.5 (5) | C12—C11—C16 | 126.6 (8) |
| N1—C1—C2 | 117.5 (6) | C11—C12—H12 | 118.4 |
| N1—C1—C6 | 117.5 (6) | C13—C12—C11 | 123.1 (8) |
| C2—C1—C6 | 125.0 (6) | C13—C12—H12 | 118.4 |
| C1—C2—H2 | 119.2 | C12—C13—H13 | 120.4 |
| C1—C2—C3 | 121.7 (7) | C12—C13—C14 | 119.2 (7) |
| C3—C2—H2 | 119.2 | C14—C13—H13 | 120.4 |
| C2—C3—H3 | 120.7 | C13—C14—H14 | 120.3 |
| C4—C3—C2 | 118.7 (6) | C15—C14—C13 | 119.3 (8) |
| C4—C3—H3 | 120.7 | C15—C14—H14 | 120.3 |
| C3—C4—H4 | 120.3 | N11—C15—H15 | 120.4 |
| C5—C4—C3 | 119.4 (7) | C14—C15—N11 | 119.3 (7) |
| C5—C4—H4 | 120.3 | C14—C15—H15 | 120.4 |
| N1—C5—C4 | 120.2 (6) | C11—C16—H16A | 109.5 |
| N1—C5—H5 | 119.9 | C11—C16—H16B | 109.5 |
| C4—C5—H5 | 119.9 | C11—C16—H16C | 109.5 |
| C1—C6—H6A | 109.5 | H16A—C16—H16B | 109.5 |
| C1—C6—H6B | 109.5 | H16A—C16—H16C | 109.5 |
| C1—C6—H6C | 109.5 | H16B—C16—H16C | 109.5 |
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2···Br1i | 0.95 | 3.06 | 3.744 (7) | 131 |
| C2—H2···Br2i | 0.95 | 3.13 | 3.884 (7) | 137 |
| C5—H5···Br1ii | 0.95 | 2.91 | 3.806 (7) | 158 |
| C6—H6A···Br1 | 0.98 | 3.03 | 3.999 (7) | 172 |
| C14—H14···Br2iii | 0.95 | 3.12 | 3.754 (8) | 126 |
| C14—H14···O1iv | 0.95 | 2.48 | 3.160 (9) | 129 |
| C15—H15···O11iv | 0.95 | 2.50 | 3.358 (8) | 150 |
| C16—H16C···O11v | 0.98 | 2.45 | 3.368 (9) | 157 |
| Symmetry codes: (i) −x+1, y−1/2, −z+1/2; (ii) x−1, y, z; (iii) x+1/2, −y+3/2, −z+1; (iv) x+1/2, −y+1/2, −z+1; (v) x−1/2, −y+1/2, −z+1. |
Acknowledgements
This work was supported by the State of Schleswig-Holstein.
References
![]() Bertini, I., Dapporto, P., Gatteschi, A. & Scozzafava, A. (1975). Inorg. Chem. 14, 1639–1643. CSD CrossRef CAS Web of Science Google Scholar
Bertini, I., Dapporto, P., Gatteschi, A. & Scozzafava, A. (1975). Inorg. Chem. 14, 1639–1643. CSD CrossRef CAS Web of Science Google Scholar
![]() Brandenburg, K. & Putz, H. (1999). DIAMOND. Crystal Impact GbR, Bonn, Germany. Google Scholar
Brandenburg, K. & Putz, H. (1999). DIAMOND. Crystal Impact GbR, Bonn, Germany. Google Scholar
![]() Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002). Acta Cryst. B58, 389–397. Web of Science CrossRef CAS IUCr Journals Google Scholar
Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002). Acta Cryst. B58, 389–397. Web of Science CrossRef CAS IUCr Journals Google Scholar
![]() Coyle, B. A. & Ibers, J. A. (1970). Inorg. Chem. 9, 767–772. CSD CrossRef CAS Web of Science Google Scholar
Coyle, B. A. & Ibers, J. A. (1970). Inorg. Chem. 9, 767–772. CSD CrossRef CAS Web of Science Google Scholar
![]() Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. Web of Science CrossRef IUCr Journals Google Scholar
Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. Web of Science CrossRef IUCr Journals Google Scholar
![]() Hussain, M. S. & Aziz Al-Hamoud, S. A. (1984). Inorg. Chim. Acta, 82, 111–117. CSD CrossRef CAS Web of Science Google Scholar
Hussain, M. S. & Aziz Al-Hamoud, S. A. (1984). Inorg. Chim. Acta, 82, 111–117. CSD CrossRef CAS Web of Science Google Scholar
![]() Kang, L., Lynch, G., Lynch, W. & Padgett, C. (2017). Acta Cryst. E73, 1434–1438. Web of Science CSD CrossRef IUCr Journals Google Scholar
Kang, L., Lynch, G., Lynch, W. & Padgett, C. (2017). Acta Cryst. E73, 1434–1438. Web of Science CSD CrossRef IUCr Journals Google Scholar
![]() Kidd, M. R., Sager, R. S. & Watson, W. H. (1967). Inorg. Chem. 6, 946–951. CSD CrossRef CAS Web of Science Google Scholar
Kidd, M. R., Sager, R. S. & Watson, W. H. (1967). Inorg. Chem. 6, 946–951. CSD CrossRef CAS Web of Science Google Scholar
![]() Lynch, S., Lynch, G., Lynch, W. E. & Padgett, C. W. (2019). Acta Cryst. E75, 1284–1290. Web of Science CSD CrossRef IUCr Journals Google Scholar
Lynch, S., Lynch, G., Lynch, W. E. & Padgett, C. W. (2019). Acta Cryst. E75, 1284–1290. Web of Science CSD CrossRef IUCr Journals Google Scholar
![]() Näther, C., Bhosekar, G. & Jess, I. (2007). Inorg. Chem. 46, 8079–8087. Web of Science PubMed Google Scholar
Näther, C., Bhosekar, G. & Jess, I. (2007). Inorg. Chem. 46, 8079–8087. Web of Science PubMed Google Scholar
![]() Näther, C., Greve, J. & Jess, I. (2002). Solid State Sci. 4, 813–820. Google Scholar
Näther, C., Greve, J. & Jess, I. (2002). Solid State Sci. 4, 813–820. Google Scholar
![]() Näther, C. & Jess, I. (2004). Eur. J. Inorg. Chem. pp. 2868–2876. Google Scholar
Näther, C. & Jess, I. (2004). Eur. J. Inorg. Chem. pp. 2868–2876. Google Scholar
![]() Näther, C., Jess, I. & Greve, J. (2001). Polyhedron, 20, 1017–1022. Web of Science CrossRef CAS Google Scholar
Näther, C., Jess, I. & Greve, J. (2001). Polyhedron, 20, 1017–1022. Web of Science CrossRef CAS Google Scholar
![]() Näther, C. & Jess, I. (2023). Acta Cryst. E79. submitted. Google Scholar
Näther, C. & Jess, I. (2023). Acta Cryst. E79. submitted. Google Scholar
![]() Neumann, T., Jess, I., Germann, L. Z., Dinnebier, R. & Näther, C. (2019). Cryst. Growth Des. 19, 1134–1143. Web of Science CSD CrossRef CAS Google Scholar
Neumann, T., Jess, I., Germann, L. Z., Dinnebier, R. & Näther, C. (2019). Cryst. Growth Des. 19, 1134–1143. Web of Science CSD CrossRef CAS Google Scholar
![]() Neumann, T., Jess, I., Pielnhofer, F. & Näther, C. (2018). Eur. J. Inorg. Chem. pp. 4972–4981. Web of Science CSD CrossRef Google Scholar
Neumann, T., Jess, I., Pielnhofer, F. & Näther, C. (2018). Eur. J. Inorg. Chem. pp. 4972–4981. Web of Science CSD CrossRef Google Scholar
![]() Parsons, S., Flack, H. D. & Wagner, T. (2013). Acta Cryst. B69, 249–259. Web of Science CSD CrossRef CAS IUCr Journals Google Scholar
Parsons, S., Flack, H. D. & Wagner, T. (2013). Acta Cryst. B69, 249–259. Web of Science CSD CrossRef CAS IUCr Journals Google Scholar
![]() Peng, R., Li, M. & Li, D. (2010). Coord. Chem. Rev. 254, 1–18. Web of Science CrossRef CAS Google Scholar
Peng, R., Li, M. & Li, D. (2010). Coord. Chem. Rev. 254, 1–18. Web of Science CrossRef CAS Google Scholar
![]() Rigaku OD (2022). CrysAlis PRO. Rigaku Oxford Diffraction, Yarnton, England. Google Scholar
Rigaku OD (2022). CrysAlis PRO. Rigaku Oxford Diffraction, Yarnton, England. Google Scholar
![]() Sager, R. S. & Watson, W. H. (1968). Inorg. Chem. 7, 2035–2040. CSD CrossRef CAS Web of Science Google Scholar
Sager, R. S. & Watson, W. H. (1968). Inorg. Chem. 7, 2035–2040. CSD CrossRef CAS Web of Science Google Scholar
![]() Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. Web of Science CrossRef CAS IUCr Journals Google Scholar
Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. Web of Science CrossRef CAS IUCr Journals Google Scholar
![]() Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8. Web of Science CrossRef IUCr Journals Google Scholar
Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8. Web of Science CrossRef IUCr Journals Google Scholar
![]() Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8. Web of Science CrossRef IUCr Journals Google Scholar
Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8. Web of Science CrossRef IUCr Journals Google Scholar
![]() Shi, J.-M., Liu, Z., Lu, J.-J. & Liu, L.-D. (2005). Acta Cryst. E61, m856–m857. Web of Science CSD CrossRef IUCr Journals Google Scholar
Shi, J.-M., Liu, Z., Lu, J.-J. & Liu, L.-D. (2005). Acta Cryst. E61, m856–m857. Web of Science CSD CrossRef IUCr Journals Google Scholar
![]() Watson, W. H. & Johnson, D. R. (1971). Inorg. Chem. 10, 1281–1288. CSD CrossRef CAS Google Scholar
Watson, W. H. & Johnson, D. R. (1971). Inorg. Chem. 10, 1281–1288. CSD CrossRef CAS Google Scholar
![]() Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925. Web of Science CrossRef CAS IUCr Journals Google Scholar
Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925. Web of Science CrossRef CAS IUCr Journals Google Scholar
This is an open-access article distributed under the terms of the Creative Commons Attribution (CC-BY) Licence, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are cited.
| CRYSTALLOGRAPHIC COMMUNICATIONS |
access



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}