

 journal menu
journal menu





issue contents
November 1998 issue
Databases for macromolecular crystallography
Proceedings of the CCP4 study weekend, January 1998
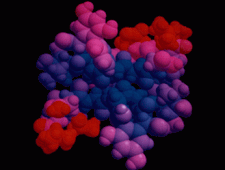
Cover illustration: The balhimycin dimer, with citrate and acetate ions in the binding pockets, colour coded on a scale where the most mobile atoms are red and the least mobile are blue. The binding pockets are clearly the most mobile, consistent with the idea that they can open and close to complex with cell-wall binding peptides. Courtesy of George Sheldrick.




research papers
Open  access
access
access
The rapid growth of the World Wide Web provides major new opportunities for distributed databases, especially in macromolecular science. A new generation of technology, based on structured documents (SD) and XML, is being developed which will integrate documents and data in a seamless manner.
Open access
access
The Iditis protein structure database provides the most comprehensive set of derived information about protein structure currently available and allows rapid searching for complex motifs.
Open access
access
The Protein Data Bank (PDB) at Brookhaven National Laboratory, a database containing experimentally determined three-dimensional structures of proteins, nucleic acids and other biological macromolecules, with approximately 8000 entries, is described.
Open access
access
A summary of macromlecular structure databases developed to date. The authors own work indicates that data are reported inconsistently and this should be addressed in the future.
Open access
access
A description is given of how the Nucleic Acid Database (NDB) is used to study nucleic acids. In addition, the way in which the technology developed by the NDB project has been extended to macromolecules in general is summarized.
Open access
access
A discussion is presented of some of the issues involved in depositing and releasing macromolecular structural information, and an outline of future directions.
Open access
access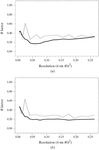
The importance of validation techniques in X-ray structure determination and their relation to refinement procedures are discussed, with particular reference to atomic resolution structures. The requirements of deposition and publication, and the role of validation tools in this are analysed. The need for a rigorously defined file format is emphasized.
Open access
access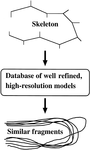
The use of databases for protein crystallographic model building, refinement, validation and analysis is reviewed, and some recent developments are discussed.
Open access
access
A description of new analytical software tools and WWW servers for studying protein sequence, structure and function is presented.
Open access
access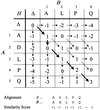
Algorithms for alignment of two or more protein sequences are described. Software for sequence analysis and database searching is summarized.
Open access
access
The Structural Classification of Proteins (SCOP) database is described. It provides a detailed and comprehensive description of the relationships of all known protein structures and can be used as a source of data to calibrate sequence search algorithms and for the generation of population statistics on protein structures.
Open access
access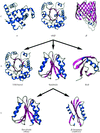
The CATH database of protein domain structures classifies structures according to their (C)lass, (A)rchitecture, (T)opology or fold and (H)omologous family. Although the protocol used is mostly automatic, manual inspection is used to check assignments at some critical stages. Described in this article is a recently established facility to search the database with the coordinates of a newly determined structure.
Open access
access
Databases of protein structural domains (DDBASE), aligned homologous protein structures (HOMSTRAD) and structurally aligned protein superfamilies (CAMPASS) are available on the WWW.
Open access
access
A survey of novel database tools for the analysis of protein–ligand interactions.
Open access
access
Analysis of data from the IsoStar library shows that many hydrophobic groups exhibit strikingly strong directional preferences in their intermolecular interactions. These directional preferences may need to be taken into account in parameterizing the next generation of protein–ligand docking programs.
Open access
access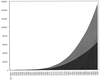
The reliability and transferability of M—L bond lengths and L—M—L bond angles from crystal structure is considered in the light of the utility of tables of `typical' bond lengths in transition-metal complexes.
Open access
access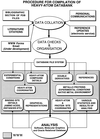
The Heavy-Atom Data Bank (HAD) described contains coordinates of heavy-atom sites derived from multiple isomorphous derivatives used in protein crystallography. HAD contains information on crystallization conditions and protein binding sites that will be of value in the preparation of heavy-atom derivatives for use in preparation of isomorphous derivatives in the method of isomorphous replacement.







