

 journal menu
journal menu





issue contents
April 2013 issue
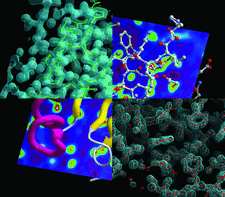
Cover illustration: A selection of model-viewing options in ArpNavigator (p. 635) depicted for PDB structure 3l9a: the C-terminal domain of a Streptococcus mutans hypothetical protein. Shown clockwise from the top left are a stick representation in solid electron density (50% opacity, 1.5σ), a ball-and-stick representation in planar density, a skeleton representation of the electron density shown as a mesh and the protein in cartoon representation in planar density.
research papers







The three-dimensional structure of an N-glycosylated and cyclic activation-associated secreted protein in a dimeric conformation corresponding to its in vivo quaternary structure has been determined at a resolution of 1.85 Å.
PDB reference: ASP-1, 4g2u
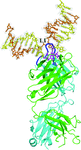
The crystal structure of a double-stranded DNA (6–4) photoproduct in complex with the anti-(6–4)-photoproduct antibody Fab was determined at 2.5 Å resolution. The T(6–4)T segment and the 5′-side adjacent adenosine are flipped out of the DNA duplex and the DNA helices are sharply kinked at the T(6–4)T segment.

The crystal structure of the N-terminal domain of V. cholerae TcpE was determined to 1.88 Å resolution revealing a monomeric six-helix bundle that is conserved in GspF family homologs. A conserved hydrophobic groove at the bottom of this domain may interact with cytoplasmic components of the pilus assembly apparatus.
PDB reference: TcpE1–102, 4hhx
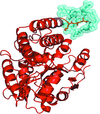
Using a carrier-protein strategy, the structure of teicoplanin bound to its bacterial cell-wall target has been determined. The structure reveals the molecular determinants of target recognition, flexibility in the antibiotic backbone and intrinsic radiation sensitivity of teicoplanin.
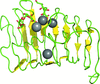
The unique active site of C. bescii family 3 pectate lyase has been structurally described, enabling a catalytic mechanism to be proposed; synergistic digestion studies suggest that PL3 can significantly improve the degradation of cellulosic biomass.
PDB reference: PL3-cat, 4ew9
Open  access
access
access
A formal derivation is provided of the 15 symmetry groups (zipper groups) available to the amyloid homosteric zipper.
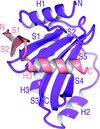
The structure of a SycH–YopH chaperone–effector complex from Yersinia reveals the bacterial state of a protein that adopts different folds in the host and pathogen environments.
PDB reference: SycH–YopH, 4gf3
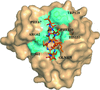
This study is the first to elucidate the structure of an enzymatically active and divergent loop-containing cyclophilin from a plant.
Open access
access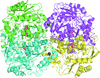
The X-ray structures of two ω-aminotransferases from P. aeruginosa and C. violaceum in complex with an inhibitor offer the first detailed insight into the structural basis of the substrate specificity of these industrially important enzymes.

Val57 point mutants of human cystatin C, which were designed to assess the influence of changes in the properties of the L1 loop on the dimerization propensity, were structurally characterized.
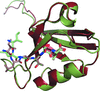
The structure of PSD-93 PDZ1 with a C-terminal GTSI extension to mimic the C-terminal tail of GluD2 shows flexibility in the binding cavity, resulting in variations in GTSI binding. Combined with the very low binding affinity of a GluD2 C-terminally derived peptide to PSD-93 PDZ1, PDZ2 and PDZ1–2, it raises doubts about the role of PSD-93 as a direct intracellular GluD2 receptor interaction partner.
PDB reference: PSD-93 PZ1ext, 4h11
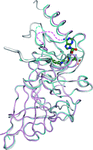
The crystal structures of histone methyltransferase SET7/9 in complexes with two previously developed AdoMet analogues revealed the structural bases for the efficient inhibition of SET7/9 by DAAM-3 and the stabilization of AAM-1, a weaker inhibitor than DAAM-3, within SET7/9 in the crystal.
Open access
access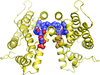
It is shown that dimerization is promoted by glycolipid binding to human GLTP. The importance of dimer flexibility in wild-type protein is manifested by point mutation that `locks' the dimer while diversifying ligand/protein adaptations.
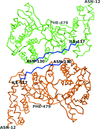
An X-ray crystallographic structure is described for unliganded Vaccinia virus poly(A) polymerase monomer (VP55), showing the first domain-level structural isoforms among either VP55, it's processivity factor VP39, or the VP55-VP39 heterodimer. The occurrence of domain-level motion specifically in monomeric VP55 is consistent with the finding that the monomeric protein undergoes saltatory translocation whereas the heterodimer does not.
PDB reference: VP55, 3owg
Open access
access
A fast analytical method for calculating mask-based bulk-solvent scale factors and overall anisotropic correction factors is introduced.
Open access
access
The molecular viewer ArpNavigator allows easy execution of ARP/wARP model-building routines while model-update steps are shown in real time, rendering the whole process transparent to the user.
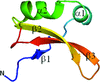
The first crystal structure of brazzein, the smallest sweet-tasting protein, has been solved at 1.8 Å resolution. The crystal structure possesses an additional α-helix that was not found in previously reported solution structures and exhibits a different molecular shape from the solution structures, with r.m.s.d.s on Cα atoms of 2.0–2.2 Å.
PDB reference: brazzein, 4he7

The crystal structure of the hydratase from Lactobacillus acidophilus (LAH) has been determined by single-wavelength anomalous dispersion. Crystal structures of apo LAH and of LAH with bound linoleic acid were refined at resolutions of 2.3 and 1.8 Å, respectively.

The crystal structure of AGME from Burkholderia thailandensis resistant to acid-induced denaturation revealed a decameric form. The in silico docking study of this enzyme with ADP-D,D-Hep supports a one-base mechanism as a major catalytic pathway.
PDB reference: ADP-L-glycero-D-manno-heptose 6-epimerase, 4ej0
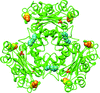
The analyses of the crystal structure of Nm23-H1 combined with H/D exchange experiments under oxidative condition revealed an intramolecular disulfide bond triggering a large conformational change that destabilizes the hexameric state and provides an environment for the oxidation of Cys109 to sulfonic acid.
PDB reference: oxidized Nm23-H1, 4eno







