

 journal menu
journal menu




issue contents
January 2022 issue

Cover illustration: CryoEM analysis of small plant biocatalysts [Dimos et al. (2022), Acta Cryst. D78, 113–123]. A micrograph of borneol dehydrogenase from Salvia rosmarinus (SrBDH1) is shown in the background while representative 2D classes encircle the cryo-EM reconstruction of tetrameric SrBDH1, with the electron density for one α-helix shown as a green mesh.
CCP4




Open  access
access
access
The implications of the AlphaFold2 protein structure-modelling software for crystallographic phasing strategies are discussed.
Open access
access
The key factors that should be considered during the planning and execution of a time-resolved crystallographic experiment are discussed, with a focus on time-resolved serial synchrotron crystallography.
research papers

Download citation



Download citation
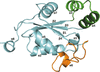
A quinary multi-hexameric helical structure of T. cruzi NDPK1 is supported by crystallographic and in vivo studies.
PDB reference: Trypanosoma cruzi nucleoside diphosphate kinase 1, 7mk0
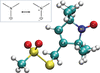
New restraints for the nitroxide radical ligand MTN defined based on quantum-chemical computations allow the accurate re-refinement of protein structures.
Open access
access
High-resolution X-ray diffraction data were collected at room temperature for light- and dark-adapted states of the archaerhodopsin-3 photoreceptor using transparent and opaque polymer-film sample mounts.
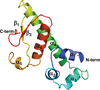
In this study, the crystal structure of anti-CRISPR-associated 2 (Aca2) was solved and it was shown that Aca2 forms dimers in solution, which are critical for promoter binding; specific residues that are critical for DNA binding and their conservation were revealed. Since the mechanism of action is largely conserved among different Aca protein families, insights are provided into their structure and function.
PDB reference: Aca2, 7ezy
Open access
access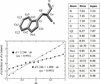
The accuracy of B factors, estimated by comparing the same atoms in numerous protein crystal structures, is rather modest: close to 9 Å2 in ambient-temperature structures and to 6 Å2 in low-temperature structures. These values are similar to those estimated two decades ago, indicating that little has changed since.
Open access
access
The binding properties of two drug-like fragments to a conformationally dynamic site in disulfide bond-forming protein A from B. pseudomallei are described.
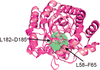
Contrary to expectation from orthologous structures from mouse and cow, a structure of holo human adenosine deaminase 1 shows that it adopts a closed conformation at the entry to its active site. This finding poses a cautionary tale for reliance on homologs to make structural inferences relevant to applications such as protein engineering or drug development.
PDB reference: human adenosine deaminase 1, 7rtg

The crystal structure of YxaL was determined, revealing an eight-bladed β-propeller fold with structural variations. The protein was subsequently engineered based on the structure, resulting in an improved plant growth-promoting activity.
Open access
access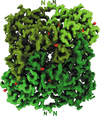
Although cryo-electron microscopy has revolutionized structural biology, its applicability to high-resolution structural analysis of comparatively small enzymes so far remains largely unexplored. Here, two cryo-EM structures of plant borneol dehydrogenases of ∼120 kDa at or below 2 Å resolution are reported.
Open access
access
It is shown that the inclusion of glycerol in single-particle cryoEM buffers does not preclude high-resolution structure determination, as demonstrated by an ∼2.3 Å resolution reconstruction of mouse apoferritin (∼500 kDa) and an ∼3.3 Å resolution reconstruction of rabbit muscle aldolase (∼160 kDa) in the presence of 20%(v/v) glycerol.
EMDB references: apoferritin in 1.7% glycerol acquired at 200 keV, EMD-24795; apoferritin in 20% glycerol acquired at 200 keV, EMD-24796; apoferritin in 20% glycerol acquired at 300 keV, EMD-24797; aldolase in 20% glycerol acquired at 200 keV, EMD-24798; aldolase in 20% glycerol acquired at 300 keV, EMD-24799







