

 journal menu
journal menu





issue contents
February 2019 issue

Cover illustration: The structure of the aromatic-amino-acid aminotransferase from Streptococcus mutans, the primary causative agent of dental caries [Cong et al. (2019), Acta Cryst. F75, 141–146].
research communications







The crystal structure of Sso0352 from Sulfolobus solfataricus, a homolog of programmed cell death 5 (PDCD5), was solved using single-wavelength anomalous diffraction. This is the first crystal structure of a PDCD5 homolog to be solved.
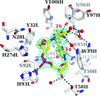
Crystal structures of the anti-(6–4) photoproduct antibody 64M-5 Fab and of its complex with dT(6–4)T indicate an induced-fit mechanism that mainly involves His93L, and also indicate that the higher affinity of 64M-5 for dT(6–4)T compared with that of 64M-2 is partially ascribable to an electrostatic interaction of His93L and a stabilization effect of Arg90L.

This communication is the first to describe the crystal structure, at 2.6 Å resolution, of a recombinant N9 influenza neuraminidase with an artificial stalk formed by the tetrabrachion (TB) tetramerization domain from Staphylothermus marinus. The structure of the N9 head without the TB domain was also determined at 2.3 Å resolution and both were compared with the N9 head derived from egg-grown virus, which was resolved to 1.4 Å resolution: the highest resolution ever reported for an uncomplexed N9 head.
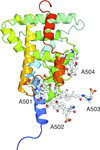
In this article, it is shown that the structure of the apo ligand-binding domain (LBD) of the retinoic X receptor, which was historically the first structure of a nuclear receptor LBD to be reported and which is still used as a reference today, contained minor inaccuracies that could be corrected using modern structure-refinement tools.
PDB reference: human RXRα, ligand-binding domain, 6hn6

A heterogeneous distribution of oligomers complicated the structure determination of the complement regulator properdin. Using TEV protease excision from recombinant oligomers, two types of properdin monomers could be crystallized.
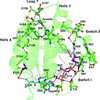
Three neutron crystallographic data sets were collected at pD 8.4, 9.0 and 9.4 from a single crystal of the small GTPase Ras. The in crystallo titration method and the crystallographic data statistics are reported. Titratable groups of interest are discussed.
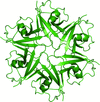
The crystal structure of a Y27W mutant of the Dishevelled2 DIX domain (DIX-Y27W) is reported at 1.64 Å resolution, which is the highest resolution to be reported for a DIX-domain structure. DIX-Y27W formed a helical polymer similar to other reported DIX domains.
PDB reference: Dishevelled2 DIX domain, Y27W mutant, 6iw3
Open  access
access
access
An automated, high-throughput crystallographic screening method is presented that has successfully been used to crystallize a panel of six diverse proteins. Iterative screening optimization is highly customizable and easy to implement for the efficient and thorough navigation of chemical space.

The rates of penetration of dyes in the molecular-weight range 250–1000 Da were investigated for a panel of dyes and a dozen different protein crystals. For cases of pure diffusion, where the dye did not appreciably bind to the protein, the rates were in the range 60–100 µm h−1 and the penetration rates approximated the actual diffusion rates. When the protein exhibited an affinity for a dye, the rates were in the range 15–30 µm h−1.

The crystal structure of the aromatic-amino-acid aminotransferase from Streptococcus mutans was solved at 2.2 Å resolution by the molecular-replacement method.
PDB reference: aromatic-amino-acid aminotransferase, 4my5
![[publBio]](/logos/publbio.gif)




