
 journal menu
journal menu


issue contents
 | STRUCTURAL BIOLOGY |
ISSN: 2059-7983
CCP4 Study Weekend 2019
Molecular replacement
Edited by Isabel Uson, Charles Ballard, Ronan Keegan and Randy Read
This virtual issue contains articles from the 2019 CCP4 Study Weekend on molecular replacement.



Open  access
access
access
Introducing the virtual special issue on the 2019 CCP4 Study Weekend on Integrated, rational molecular replacement at https://journals.iucr.org/special_issues/2020/CCP42019/.
Open access
access
The employment of directed acyclic graphs to advance the tracking, control and appraisal of crystallographic phasing strategies is discussed.

Open access
access
Local error estimates can be used to improve the success of models in molecular replacement.
Open access
access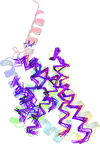
Predicted ab initio protein models from online databases are a useful supplement to the PDB for molecular replacement, but usually require nontrivial processing to succeed.
Open access
access
Improvements to the sensitivity of the search for suitable molecular-replacement search models in SIMBAD through the use of Phaser and an ensemble-based model database are reported.
Open access
access
ALEPH characterizes the main-chain geometry of small, noncontinuous fragments to flexibly annotate secondary structure, decompose folds, extract libraries and superpose fragments. Secondary and tertiary structure are described through networks of characteristic vectors, which are defined between the centroids of the Cα and carbonyl O atoms in a peptide.

Download citation



Download citation
Open access
access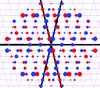
Examples are presented of successful data-processing, phasing and refinement strategies for non-merohedral twins, covering the range from minerals to proteins.
Open access
access
Translational noncrystallographic symmetry (TNCS) was analysed using a curated database of 80 000 protein structures to inform an algorithm for the detection of TNCS order.
Open access
access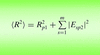
An automatic pipeline based on molecular-replacement phases is described for the automatic crystal structure solution of protein and DNA/RNA molecules.
Open access
access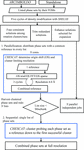
The software ALIXE, which is available as a standalone program as well as integrated into the ARCIMBOLDO programs, combines partial solutions in reciprocal space to increase the signal and reduce the number of intermediate solutions. Its new implementation is faster and can be used by default in the process of phasing with fragments even on modest hardware.
Open access
access
When phasing cannot be accomplished from a partial polyalanine starting model, extending the model with side chains in a multi-solution way may succeed. SEQUENCE SLIDER implements this approach for use in ARCIMBOLDO.
Open access
access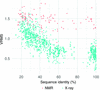
Improved coordinate error estimates are proposed for the X-ray and NMR models used for maximum-likelihood-based molecular-replacement phasing.
Open access
access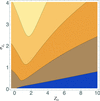
The information content gained by making a diffraction-intensity measurement is a natural criterion for deciding which data make a useful contribution and which can legitimately be omitted from a calculation.
Open access
access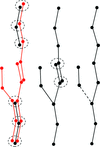
A novel method for the enhancement of automated protein model building using polypeptide fragments of homologous structures is presented.
Open access
access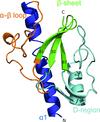
A practical perspective on molecular-replacement (MR) structure-determination pipelines is presented, using PilA1 from Clostridioides difficile as an example of a difficult case. A manual approach informed by the biology of the system under study is described, together with ab initio MR structure determination.
PDB reference: PilA1, 6t8s
Open access
access
Fragment-based determination of a proteinase K structure from MicroED data using ARCIMBOLDO_SHREDDER
A 1.6 Å resolution MicroED data set of proteinase K is phased using fragments derived from distantly related sequence homologues. ARCIMBOLDO_SHREDDER expands the phasing options for MicroED applications, overcoming the need for complete and highly accurate search models.
PDB reference: proteinase K, 6v8r
Open access
access
A partial 30% atomic model from a 7.8 Å resolution cryo-EM volume was used to phase X-ray data by molecular replacement.
Open access
access
The solution of coiled-coil crystal structures may be achieved by AMPLE through the use of ensembled ab initio models in molecular replacement. Improvements in ab initio modelling of elongated helices and oligomeric coiled-coils allow AMPLE to solve a greater number of coiled-coil structures and at lower resolution than previously achieved.







