journal menu
journal menu


issue contents
 | STRUCTURAL SCIENCE CRYSTAL ENGINEERING MATERIALS |
ISSN: 2052-5206
Special issue on quantum crystallography
Guest editors: Jean-Michel Gillet and Piero Macchi
This virtual issue collects together articles from QCrOM2020.

Cover illustration: The authors contributing to this special issue all participated in the first quantum crystallography meeting (QCrOM2020) held online in August 2020. The issue brings together articles originally published in Acta Crystallographica Section B between June 2021 and December 2021.



Free 
An overview is given by the guest editors of the papers published in the virtual special issue on quantum crystallography.
Open access
access
This work presents a detailed analysis of the recently published deformation potentials for application in orbital-free density functional theory, that are able to take the interaction between atoms into account through the help of their electron densities only. It is shown that the present ansatz provides a systematic pathway beyond the recently introduced atomic fragment approach.


The electrostatic, exchange, Weizsacker and Pauli potentials, as well as associated partial electron densities, reconstructed from experimental electron density using the orbital-free density functional theory formalism, are applied for the bonding analysis for a heteromolecular crystal of ammonium hydrooxalate oxalic acid dihydrate.

Download citation



Download citation
Experimental charge density is applied for covalent and noncovalent bond characterization in Appel's salt (4,5-dichloro-l,2,3-dithiazolium chloride). The chalcogen and halogen bonds are categorized using the gradient fields of electron density and electrostatic potential.
CCDC reference: 2088696
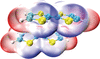
High-level quantum chemical calculations provide quantitative insights into the nature of intermolecular interactions in the stacking systems of tetrathiafulvalene and selected derivatives.
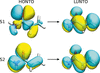
X-ray restrained extremely localized molecular orbitals (XR-ELMOs) were used in QM/ELMO calculations of excited states to describe the environment of a chromophore. It is demonstrated that the XR-ELMOs can be profitably used for this purpose, also confirming the capability of the X-ray restrained wavefunction approach in capturing environmental effects on the electron density.
Open access
access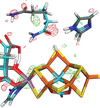
Quantum refinement has been shown to be a powerful approach to interpret and improve macromolecular crystal structures. Previous studies have shown that the results of quantum refinement can be improved if the charge of the quantum mechanical (QM) system is reduced by adding neutralizing groups. Here it is shown that a similar improvement can be obtained if the original highly charged QM system is instead immersed in a continuum solvent in the QM calculations.
Open access
access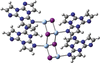
The different nature of the emissive states of AgI and CuI one- and three-dimensional coordination polymers is here explained by a higher covalent character of the Cu—N bond with respect to the Ag—N one.
Download citation
Download citation
Open access
access
Intermolecular interactions of the new drug candidate sila-ibuprofen are investigated in the crystal and in the enzyme cyclooxygenase-II.
This article describes the joint refinement program MOLLYNX dedicated to spin-resolved electron density distribution. Multipole and atomic orbital models are described and applications for organic and inorganic magnetic materials are discussed.
Download citation
Download citation
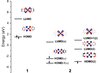
Differences between the formal oxidation states of two nickel(II/III) coordination compounds with a non-innocent ligand were studied. There are differences between the theoretical calculations as well as between various experimental results.
The influence of data manipulation on X-ray wavefunction refinement is analysed. While the Hirshfeld atom refinement always reaches the same results, X-ray constrained wavefunction fitting is extremely sensitive to crystallographic data manipulation as a consequence of the variability of the experimental uncertainties for different resolution intervals.
This article retraces different methods that have been explored to account for the atomic thermal motion in the reconstruction of N-representable one-electron reduced density matrices from experimental X-ray structure factors and directional Compton profiles.
Download citation
Download citation
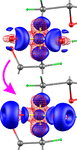
The transfer of theoretical multipole parameters to the experimental model was found to be helpful in separating the static electron density from the contaminant smearing effects emerging due to insufficiently accounted for atomic motion. A special refinement procedure resulted in significant improvement of the static electron density.
CCDC reference: 2084659