
 journal menu
journal menu



issue contents
 | STRUCTURAL BIOLOGY |
ISSN: 2059-7983
CCP4 Study Weekend 2022
Current trends in macromolecular model refinement and validation
Edited by Melanie Vollmar, Rob Nicholls and Svetlana Antonyuk
This virtual issue contains articles from the 2022 CCP4 Study Weekend.





Open  access
access
access
The Guest Editors introduce the special issue based on talks at the CCP4 Study Weekend 2022. The virtual issue is available at https://journals.iucr.org/special_issues/2023/CCP42022/.
Open access
access
Errors from measurement, data processing and modelling are present throughout structures deposited in the Protein Data Bank. Identifying them is only the first step. What needs to change in order to minimize their impact?
Open access
access
A new protein structure validation method using hydrogen-bonding parameters is described.

Open access
access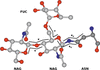
The Privateer software allows structural biologists to evaluate and improve the atomic structures of carbohydrates, including N-glycans. This software has recently been extended to check glycan composition through the use of glycomics data, and the broadening of its scope is presented in this article.
Open access
access
This article describes the Collaborative Computational Project No. 4 (CCP4). It is intended as a general literature citation for the use of the CCP4 software suite in structure determination.
Open access
access
The paper describes CCP4 Cloud, an online system for macromolecular structure determination based on the CCP4 software suite.
Open access
access
Alternative conformations are underrepresented in current PDB models due to difficulties in manually detecting, building and inspecting multiple conformers. To overcome this shortcoming, an automated multi-conformer modeling program, FLEXR, has been developed that uses Ringer-based electron-density sampling to explicitly build multi-conformer models for refinement.
Open access
access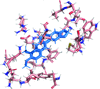
In situ quantum-mechanical optimization of ligand structures provides accurate ligand restraints for macromolecular refinement.
Open access
access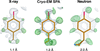
The macromolecular refinement package REFMAC5 from the CCP4 suite has been extended by the incorporation of algorithms for neutron crystallography.
Open access
access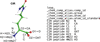
The restraint-generation part of the macromolecular atomic model-refinement program REFMAC5 has been delegated to GEMMI. A controller program was implemented in Servalcat to distribute tasks between GEMMI and REFMAC5.
Open access
access
The improved joint X-ray and neutron refinement procedure in Phenix optimizes two different models against the X-ray and neutron data sets. This approach is shown to reduce overfitting compared with refining the models separately.
Open access
access
It is found that research mentoring of undergraduate students can result in meaningful contributions to scientific disciplines. Here, an outcome-oriented, skill-based mentoring strategy is proposed and evaluated in the context of the CASP14 community challenge.
Open access
access
Defining best practice in science is challenging. International consensus is facilitated by the International Science Council via its members such as the International Union of Crystallography. The IUCr Journals editorial boards are a practical forum for setting the criteria to decide if a study's files are truly the `version of record'. Within that, reality involves a variance of reasonable workflows. Workflows must be detailed carefully by authors in explaining what they have done.
Open access
access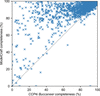
ModelCraft is a new model-building pipeline which improves on previous Buccaneer pipelines by the addition of new steps that make it more likely to build a complete protein model, especially when starting from a poor molecular-replacement solution.
Open access
access
The explicit refinement of Ramachandran, rotamer and clash criteria at now-prevalent lower resolutions (2.5–4 Å) has made the current, traditional model validation at the Protein Data Bank nearly useless in this range, since quite poor structures can have perfect scores. Fortunately, new criteria and programs such as ISOLDE, CaBLAM and AlphaFold are coming to the rescue, are already very useful and should be extensible into an effective new community standard.
Open access
access
The use of AlphaFold2 predictions for the detection and correction of sequence-register errors among protein structures determined using cryo-EM deposited in the Protein Data Bank is described.
Open access
access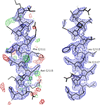
It is shown that checkMySequence, an automated method for validating sequence assignment in cryo-EM structures of proteins, can be used to validate crystal structure models.
Open access
access
The use of predicted models for macromolecular structure determination in CCP4 is discussed.
Open access
access
The accuracy of the models predicted using AlphaFold and RoseTTAFold eases phasing with molecular replacement and raises the question of model bias. ARCIMBOLDO_SHREDDER solves structures while aiming to establish the experimental information in a crystallographic determination by introducing systematic, local verification of phasing solutions.

Open access
access
phenix.process_predicted_model and ISOLDE provide seamless integration of AlphaFold-predicted models into protein structure-solution workflows.







