journal menu
journal menu




issue contents
February 1996 issue
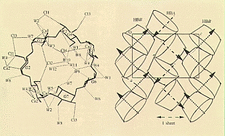
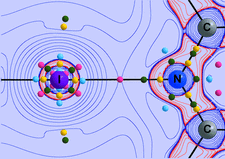
Cover illustration: Molecular unit of ![[beta]](/logos/entities/beta_rmgif.gif) -cyclodextrin (left), showing interactions which form antiparallel herringbone chains (right). Courtesy
of I. Nicolis, A. W. Coleman, P. Charpin and C. de Rango.
-cyclodextrin (left), showing interactions which form antiparallel herringbone chains (right). Courtesy
of I. Nicolis, A. W. Coleman, P. Charpin and C. de Rango.
international union of crystallography



Free 

research papers

A modification of the present bond-valence model concept is suggested for compounds with SbIII-X bonds (X = S, Se), which considers the influence of the lone-electron pair of Sb. A method to calculate a non-integer effective valence of SbIII from the stereochemistry of ψ-[SbIIIXn] polyhedra is derived.

The angles of tilt of the BX6 octahedra in orthorhombic perovskites, ABX3, are directly related to the ratio of the volumes of AX12 and BX6 polyhedra. This relationship is investigated for known structures of oxides, fluorides and hydrides.

Download citation




Download citation

The structure of the ζ-phase in the Pu-U system has been determined by neutron powder diffraction. The phase is rhombohedral with 58 atoms in the primitive unit cell.
Download citation
Download citation

A refinement was performed on single-crystal X-ray diffraction data in a ninefold supercell. The monoclinic symmetry corresponds to a systematic shift of the modulation waves of consecutive slabs by π/9 with respect to orthorhombic Bi2Sr2Ca-Cu2O8+δ. Additional oxygen sites were detected in the Bi-O chains.

A trial structure determination is described based on the initial assumption of tetrahedral close-packing. Only space groups, unit-cell dimensions and metallic radii are needed for determination.

Combined neutron and X-ray diffuse data were used to analyse the static disorder in CaO-stabilized `cubic' zirconia.

Diffuse neutron as well as X-ray intensities of calcium-stabilized zirconia can be described by a correlated distribution of the two types of microdomains. An anomolous intensity drop between 1250 and 1300 K may be related to a loss of correlated domains and the formation of free oxygen vacancies.
Download citation
Download citation

Pb5Al3F19 undergoes a phase transition from ferroelectric to antiferroelectric at 270 K on heating. The atomic displacements from the ferroelectric structure to a hypothetical paraelectric structure led to a predicted Curie temperature of 860 (230) K.

A method for the calculation of the point in a coordination polyhedron with minimal variation of distances to the vertices is described. Its application in the analysis of irregular coordination polyhedra is proposed.
Download citation
Download citation

Modeling of the H-atom thermal motion by anharmonic coefficients and by a three-site split-atom model. Comparison of interlayer interactions in Mg(OH)2 and Ca(OH)2.

The crystal structures of four hydroxylapatite samples of different magnesium content have been investigated by X-ray powder pattern fitting. The results of the different refinement procedures indicate that apatite structure can host at most 10 Mg-atom-%, which is preferentially located in one of the two crystallographically independent sites.
Download citation
Download citation

The structure of Nd2CuO4 at 20 K has been refined and compared with the room-temperature structure. The Gram-Charlier temperature-factor formalism was used to describe the Nd-atom displacements.

The incommensurately modulated structure of TaGe0.354Te2 was determined by single-crystal X-ray diffraction, using crenel functions in the refinement in combination with an orthogonalization procedure.
Download citation
Download citation

α-Ferrocenyl alcohols, FcCH(OH)R and FcCPh(OH)R, exhibit a range of hydrogen-bonding patterns, including the formation of spiral chains and closed R22(4) dimers, as well as monomers containing intramolecular O-H⋯π(C5H5) interactions.
Download citation
Download citation

The presence of a Ca2+ salt in a β-cyclodextrin lattice is characterized by direct interactions of the cations with cyclodextrins and no contacts between cations and anions.
CCDC reference: 131873
Download citation
Download citation

The p-dichlorobenzene (pdcb)/H-ZSM-5 single crystal studied contains 2.56 (2) pdcb molecules per unit cell at the intersection of channels. Energy calculations support this interpretation.
CCDC reference: 131868
Download citation
Download citation

The zeolite H-ZSM-5 has been loaded with eight molecules of p-dichlorobenzene per unit cell. The structural details are comparable to those in the high-loaded H-ZSM-5/8p-xylene complex.
CCDC reference: 131869
Download citation
Download citation
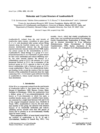
Azadirachtin-H, unlike other members of this family of C-seco-limonoids, has no carbomethoxy group at C(11), has a cyclic hemiacetal function at C(11) and has stereochemistry at C(11) opposite to that reported earlier based on NMR data. The molecule crystallizes in space group I4 with disordered ethyl acetate solvate.
CCDC reference: 131861
Download citation
Download citation

The effect of 1,5-disubstitution on the photochemistry of 9,10-ethenoanthracene-11,12-diesters has been studied and correlated with the crystal structures determined for several derivatives. Relationships are sought between the ratios of the two regioisomeric photoproducts and molecular and crystal structures; both electronic and steric effects are considered, but no one effect seems to dominate.
Download citation
Download citation

The electrostatic properties and the nature of the electron charge transfer of the m-nitrophenol molecule in the orthorhombic structure are discussed.
CCDC reference: 131874

This paper describes a novel method for predicting the crystal structure of organic molecular materials which employs a series of successive approximations to focus on structures of high probability, without resorting to brute force search and energy minimization of all possible structures.
Download citation
Download citation

Conformationally dependent changes of the geometry of choline esters are elucidated based on results obtained from X-ray crystallographic studies and molecular mechanics calculations.
Download citation
Download citation

The structures of three diphenylamine-containing crowns are reported and compared. An interesting substitutional disorder due to conformational flexibility is analysed.

Polymorphism and computer generation of crystal structure from molecular structure for organic compounds are analysed.
Download citation
Download citation

The title compound crystallizes into a layered structure containing two-dimensional pseudohexagonal hydrogen-bonded networks.
CCDC reference: 131860