journal menu
journal menu




issue contents
June 1996 issue

Cover illustration: View of 1,25-dihydroxycholecalciferol structure along molecular channels filled with water molecules. Courtesy of K. Suwinska and A. Kutner.
research papers




The composite crystal (SbS)1.15(TiS2)2 was prepared, which is built up of alternatingly interface-modulated SbS double layers and paired sandwiches of (TiS2)2.

The structure of (La0.95Se)1.21VSe2, a misfit layer chalcogenide, was determined. It is built of an alternating stack of LaSe double layers, which contain La vacancies, and VSe2 sandwiches.

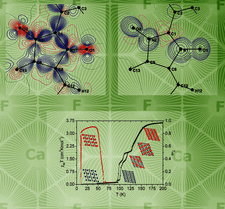
The deformation electron density (Δρ) has been determined for synthetic samarium orthoferrite, SmFeO3, using focused λ = 0.7 Å synchrotron X-radiation. The approximate Δρ map symmetry indicates that the electron density is perturbed significantly by the cation-cation interactions.

Download citation




Download citation

Deformation electron densities for synthetic C-type Y2O3, Dy2O3 and Ho2O3 were determined using focused synchrotron (λ = 0.7 Å) and Mo Kα (λ = 0.71073 Å) X-radiation. Approximate symmetry in the Δρ maps indicates that the cations deform more strongly by interaction with other cations than with O atoms, unless the symmetry of the O-defined crystal field is low.
Download citation
Download citation

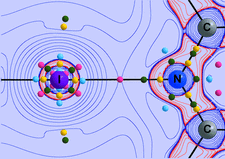
ynchrotron X-ray and neutron powder diffraction experiments on M(OH)I (M = Sr, Ba). The longest H⋯Y distances (Y = Cl, Br, I), for which hydrogen bonds have been established, are 2.80, 2.90 and 3.25 Å, respectively.
Download citation
Download citation

High-resolution synchrotron powder diffraction reveals a monoclinic distortion in β-Fe2PO5 at room temperature. A Rietveld refinement is performed and the description of the structure shows that the two iron sites are very similar.
Download citation
Download citation

Changes in the ferroelectric transition temperature in PbBi2-Nb2O9 have been shown by Raman and neutron diffraction studies to relate to Pb-Bi ordering.
Download citation
Download citation

The crystal structures of both polymorphs of NaCoPO4 have been determined by single crystal X-ray diffraction. Bond-valence analysis reveals that the polymorphism is due in part to the bonding around the Na atoms.
Download citation
Download citation

Structural parameters and their thermal changes in magnetite have been studied and the following have been observed: the oxygen coordinate remained almost constant at temperatures below ca 600 K, but it began to increase above ca 600 K; the thermal vibrational ellipsoid at the B site was prolate and oblate at temperatures below and above 630 K, respectively.
Download citation
Download citation

Charge density studies of NH4[Ti(C2O4)2].2H2O are presented using Mo Kα and Ag Kα radiation. The asphericity in density around Ti and the bonding feature of C2O42− as well as hydrogen bonding are clearly demonstrated.
Download citation
Download citation

Inversion twinning was observed in crystals of (η5-cyclopenta-dienyl)(η4-tribenzylidenediide)dimethyltantalum; the molecules have threefold symmetry imposed on them, which also requires that they will be disordered. A satisfactory model was developed and refined, but the problems of data reduction with inversion twinning present were not completely resolved.
CCDC reference: 131572
Download citation
Download citation

Topological analysis of the experimental and theoretical electron density of lithium bis(tetramethylammonium) hexanitrocobaltate(III) crystals sheds light on the nature of the ligand-transition metal interaction and on the asphericity of nonbonding 3d-electrons around the Co atom.

The use of anomalous dispersion, including synchrotron radiation, to determine the metal-atom substitution sites for the test case of NiAPO is described. Optimization of the experimental protocols is discussed for cases of partial occupancy and for more than one metal atom of similar atomic number.
Download citation
Download citation

The crystal structures of five 1,4-dihydro-2,3-quinoxalinedione antagonists of the NMDA modulatory glycine binding site have been determined and the hydrogen-bonding graph sets of all quinoxalinedione crystal structures have been analyzed. Two types of hydrogen-bonding motifs are found: a centrosymmetric dimeric ring and an infinite chain; this analysis suggests biological activity requires one donor and two hydrogen-bond acceptors for receptor recognition.
Download citation
Download citation

The X-ray structure and molecular modelling of 1,5-bis-(dimethylamino)-1,5-diphenylpentadienylium perchlorate are studied. Modelling of two other related cyanine dyes reproduces the solid state structure with good accuracy.
CCDC reference: 131583
Download citation
Download citation

The crystal and molecular structures of isomers 1 and 2 of 4-methyl-1,3,2-dioxathiane 2-oxide and 1,3,2-dioxathiepane 2-oxide have been determined by single crystal X-ray diffraction methods at low temperatures.
Download citation
Download citation

A systematic structural analysis of serotonin receptor ligands has identified their similarities with the endogenous ligand serotonin (5-hydroxytryptamine or 5-HT) and the stereochemical differences which determine selectivity for the various receptor subtypes: 5-HT1, 5-HT2 and 5-HT3.
Download citation
Download citation

(X-X) and X-(X + N) charge densities of LAP are compared together with the corresponding topology and electrostatic potentials. Reliable results may be obtained from (X-X) data only.

Conceptual clustering, a machine learning technique, is applied to the determination of conformational preferences from crystal structure data.
Download citation
Download citation

The solid-state conformation of the biologically active form of vitamin D3 is discussed.
CCDC reference: 131584
Download citation
Download citation

Two modifications of 4-amidinoindanone guanylhydrazone have been elucidated. The generality of a combination of high-precision computational chemistry and Rietveld refinement is demonstrated for one modification where no single crystals were available.
Download citation
Download citation

Two crystal structures of benzene-1,2-disulfonic acid salts have been determined and the cogging of the sulfonate groups as well as the possible pathways of conformational isomerization of the dianion are discussed, based on a PM3 enthalpy hypersurface and compared with known molecules containing adjacent threefold substituents -XY3.