journal menu
journal menu




issue contents
December 2004 issue
Model building and refinement
Proceedings of the CCP4 study weekend

Cover illustration: This is a Kendrew wire model of alcohol dehydrogenase that is about to undergo a round of rebuilding by Maelle Cambillau (p. 2115).







research papers
Open  access
access
access
A review of the author's computer-graphics developments is presented, as well as a description of a secondary-structure template-building system that works in conjunction with a new sequence-decoration scheme.
Open access
access
Coot provides both a set of libraries and a stand-alone application for model building and map fitting using molecular graphics.
Open access
access
An overview of statistical pattern-recognition techniques is given with a bias towards crystallographic applications, especially automated model building.
Open access
access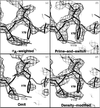
Prime-and-switch phasing can reduce atomic model bias in model-based electron-density maps.
Open access
access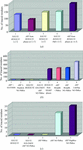
A few examples are presented on the use of different programs to achieve the goal of structure solution and the associated practicalities that can make the difference between solving or not solving a structure
Open access
access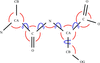
A general introduction to the models and methods used in contemporary macromolecular refinement programs.
Open access
access
An explanation of maximum likelihood in macromolecular crystallography.

Download citation


Download citation
Open access
access
The organization and use of a dictionary of monomers and links for restrained macromolecular refinement with application in the program REFMAC5 is described.
Open access
access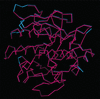
A multivariate likelihood function that directly and dynamically incorporates experimental phase information in model refinement leads to an automatically built model when current functions fail.
Download citation
Download citation
Open access
access
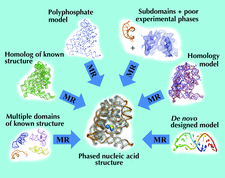
The N-particle modelling method of conditional optimization is discussed. Its model-building potential at medium to low (2.4–3.0 Å) resolution is compared with ARP/wARP and RESOLVE and preliminary results indicate its potential for phasing by ab initio modelling.
Open access
access
BUSTER–TNT is a maximum-likelihood macromolecular refinement package. BUSTER assembles the structural model, scales observed and calculated structure-factor amplitudes and computes the model likelihood, whilst TNT handles the stereochemistry and NCS restraints/constraints and shifts the atomic coordinates, B factors and occupancies. In real space, in addition to the traditional atomic and bulk-solvent models, BUSTER models the unbuilt parts of the structure through low-resolution probability distributions for the random positions of the missing atoms.
Open access
access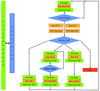
The new ARP/wARP control system, rebuilding and validation modules are described.
Open access
access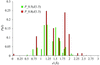
Algorithms and scoring functions for automated interpretation of electron density of protein-bound ligands are described.
Open access
access
Details of the Uppsala Electron Density Server (EDS; https://eds.bmc.uu.se/) are described, including the methods and programs that are used to calculate the maps and the files and information that are made available through the server.
Open access
access
Description of the new CCP4 Coordinate Library.
Open access
access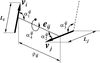
A description of a new tool for the comparison of protein structures in three dimensions and analysis of structure similarity scores.
Open access
access
The identification of the four structural domains of Src kinase by iterative analysis of error-scaled difference distance matrices is described.
Open access
access
Genetic algorithms have been implemented for real-space optimizations in a new fitting program, MIfit, for real-space refinement of protein models and heavy-atom searches. Some programming tips and examples are presented to aid others who might want to use genetic algorithms in their own work.
Open access
access
A public web-based facility to infer, analyse and graphically represent the likely modes of a protein motion, starting from a static structure, is presented.
Open access
access
The CCP4 molecular-graphics project will provide a wide range of graphics-related functionality for crystallography and related disciplines. The current status and future plans are presented.