journal menu
journal menu




issue contents
February 2011 issue
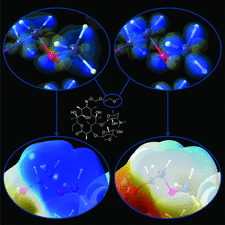
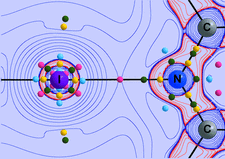
Cover illustration: The cover picture highlights an artefact in the electrostatic potential of the macrolide antibiotic roxithromycin as derived via multipole refinement of X-ray diffraction data. Ignoring the hydrogen disorder (left) in the oxime chain leads to a falsely positive electrostatic potential there as a result of the multipole model's being flexible enough to fit the averaged electron density. If the invariom formalism, which uses theoretical electron density, is applied to the disordered region (right) the artefact disappears. A procedure for obtaining reliable electron density distributions in the presence of disorder has been proposed [Holstein et al. (2010). Acta Cryst. B66, 568-577].
research papers



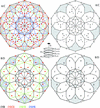
The building principles of Al-based decagonal quasicrystals and their approximants are discussed from a geometrical point of view.

Download citation




Download citation
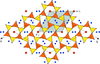
The modulated structure of natural nepheline has been determined using the superspace approach.
Download citation
Download citation
The crystal structures of two new high-pressure AlPO4 phases synthesized at 6–7 GPa and 1523–1773 K are reported. Both phases contain doubly bent chains composed of six edge-shared Al polyhedra linked by PO4 tetrahedra.
Download citation
Download citation
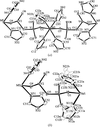
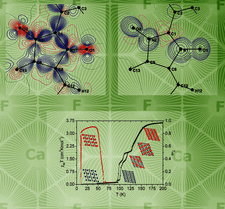
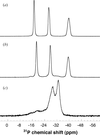
The polymeric isomorphous hybrid inorganic–organic vanadium oxide compounds [M(Im)4V2O6]∞, M = Mn, Co, Ni, Im = imidazole, were investigated between 100 (2) and 295 (2) K by single-crystal X-ray diffraction. The compounds contain two-dimensional polymeric sheets packed perpendicularly to c* and undergo a reversible order–disorder phase transition. The room-temperature P42/n disordered phase (Z = 8) is reversibly transformed to the I41/a ordered phase (Z = 32) below 281 K for the Mn and 175 K for the Co compounds, requiring a change of the hydrogen-bond connectivity for two of the eight imidazoles per asymmetric unit of the I41/a structure. The ordered phase of the Ni compound was found to have the space group P2/n (Z = 8) at 100 K. Models for the phase-transition mechanisms are considered.
Download citation
Download citation
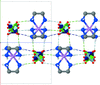
The structures of the isostructural title compounds have been determined at multiple temperatures. The disorder in the cation due to hydrogen bonding and/or ring puckering and the Jahn–Teller effect were analyzed using a rigid-body model and normal coordinate analysis.
Download citation
Download citation
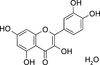
A new crystal structure of quercetin monohydrate determined by X-ray diffraction is described. A transferred aspherical atom model is used for electron-density modelling. The resulting electron-density distribution and derived quantities are compared with the theoretical multipolar atom model.
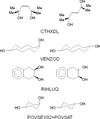
A list of stoichiometric, ordered, molecular co-crystals of isomers, near isomers and molecules that are almost the same has been compiled in order to understand better the circumstances under which fractional crystallization is likely to fail.
There is excellent agreement between torsional distributions from crystal structure data and torsional energy profiles computed using DFT for a range of π-acceptor substituents attached to cyclopropane. Asymmetry in ring bond lengths is also highly comparable in the crystal structure and computational results.
international union of crystallography
Free