journal menu
journal menu




issue contents
October 2022 issue

Cover illustration: The Pca21 structure of 1,3-di(tertbutyl) cyclopentadienyl pentaphosphaferrocene viewed down [010]. Dotted curves mark the interface between layers. [See Stöger et al. (2022). Acta Cryst. B78, 734–744.]
scientific commentaries



Free 
A short commentary is given on a recent paper by Carolyn Pratt Brock [Acta Cryst. B78, 576–588] which discusses the occurrences of approximate symmetries in space group P1.
research papers
Open access
access
Using first-principles density functional theory, the electronic and optical properties of monolayer and multilayer nanosheets of molybdenum carbon fluoride (Mo2CF2), a two-dimensional (2D) transition-metal carbide MXene, were investigated. The unique behavior of its optical properties along with the ability to control its electronic and optical properties enhances the potential of 2D Mo2CF2 for various applications in the fields of electronics and energy storage.
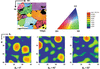
The crystallographic texture evolution in 3xxx Al alloys in different processing routes is presented and compared to the anisotropic effects occurring during deep drawing. The earing phenomena in Al alloys are correlated to the produced texture and therefore their understanding is crucial in order to highlight the importance of texture control throughout the thermomechanical process.
Open access
access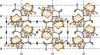
The diffuse scattering of 1,3-di(tert-butyl)cyclopentadienyl pentaphosphaferrocene (Cp′′FeP5) is modelled with closed-form expressions derived from a growth model with the range of interactions (reichweite) s = 2.

Download citation




Download citation
Open access
access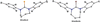
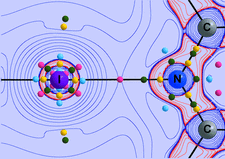
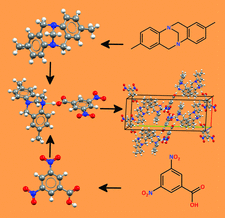
Utilizing the N-heterocyclic chalcogenones hexahydro-1,3-bis(2,4,6-trimethylphenyl)-2H-1,3-diazepine-2-thione (SDiazMesS) and hexahydro-1,3-bis(2,4,6-trimethylphenyl)-2H-1,3-diazepine-2-selone (SDiazMesSe) as halogen-bond acceptors, 24 new cocrystals were prepared. The solid-state structures of the parent molecules were also determined, along with those of their acetonitrile solvates.
Download citation
Download citation

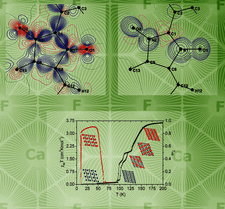
The effect of hydrostatic compression on the crystal structure of glycinium phosphite is explored by single-crystal X-ray diffraction and DFT calculations and discussed in relation to a possible ferroelectric phase transition.
Download citation
Download citation
Open access
access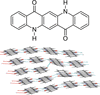
Various lattice defects in the αI-phase of quinacridone (C20H12N2O2) were simulated using lattice-energy minimizations, including vacancies, stacking faults, screw and edge dislocations, twinning and orientational faults. Twinning and orientational faults of entire chains were calculated to occur most frequently.

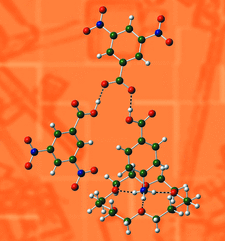
The limits of DFT calculations for distinguishing between salt and cocrystal are shown. Issues related to false salt identification mentioned in other articles are confirmed. A workflow was developed which, by the use of computational methods, allows the confirmation of salt, cocrystal and intermediate salt-cocrystal phases as well as identifying structures for which the confirmation cannot be made and further analysis is required.
Download citation
Download citation
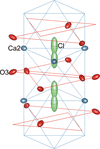
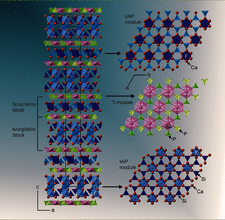
Apatite-type compounds Ca5(VO4)3Cl and Ca4.78 (1)Na0.22(PO4)3Cl0.78 were synthesized and their structures examined with single-crystal X-ray diffraction. The position of the channel anion in Ca5(VO4)3Cl was split into two. Dynamic disordering of Cl− between these two positions was suggested from their highly anisotropic displacements (dominant in 〈001〉) with saddle-shaped electron density distribution through the regular Ca2 triangle.