

 journal menu
journal menu





issue contents
May 2011 issue
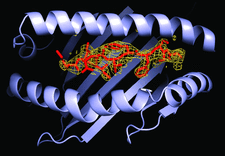
Cover illustration: The crystal structure of HLA-A3 in complex with a ninemer peptide. The view is into the binding groove, with the peptide model in red and the corresponding electron density in yellow chicken wire (p. 447).
research papers






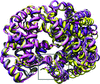
This study presents the crystal structure of Greyhound hemoglobin (GrHb) determined to 1.9 Å resolution.
PDB reference: Greyhound hemoglobin, 3pel
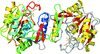
Two structure determinations of M. smegmatis IspD are reported: one in complex with CTP and the other in complex with CMP. In addition, the enzymatic characterization of the M. tuberculosis homologue and the structure of this enzyme in complex with CTP are reported.
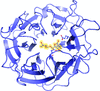
Crystal structures of the exo-α-1,5-L-arabinanase Abnx from P. chrysogenum 31B and of its complex with arabinobiose were determined at high resolutions of 1.14 and 1.04 Å, respectively.
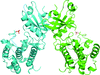
The endoplasmic reticulum-localized transmembrane kinase PERK is one of three major ER stress transducers. The crystal structure of PERK's kinase domain has been determined to 2.8 Å resolution.
PDB reference: PERK kinase domain, 3qd2
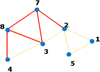
Network representation of protein structures and analysis in terms of network parameters provide efficient tools to capture conformational changes in atomistic detail in a global milieu.
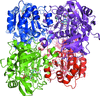
The X-ray crystal structure and a small-angle X-ray scattering solution structure of sheep liver sorbitol dehydrogenase have been determined. The details of the interactions that enable the tetramer scaffold to be the functional biological unit have been analyzed.
PDB reference: sheep liver sorbitol dehydrogenase, 3qe3
Open  access
access
access
The structure of the human major histocompatability (MHC) class I molecule HLA-A*0301 (HLA-A3) in complex with a nonameric peptide (KLIETYFSK) has been determined by X-ray crystallography to 2.7 Å resolution.
PDB reference: HLA-A*0301 complex, 2xpg
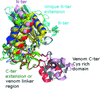
The first structure of a two-CAP-domain protein, Na-ASP-1, from the major human hookworm parasite N. americanus refined to a resolution limit of 2.2 Å is presented.
PDB reference: Na-ASP-1, 3nt8
Open access
access
Pi sampling, derived from the incomplete factorial approach, is an effort to maximize the diversity of macromolecular crystallization conditions and to facilitate the preparation of 96-condition initial screens.
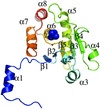
The X-ray crystal structure of human dual-specificity phosphatase 27 (DUSP27) is reported at 2.38 Å resolution.
PDB reference: DUSP27, 2y96
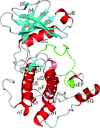
AMP-activated protein kinase (AMPK) functions as a sensor to maintain energy balance and is therefore a potential target for drug design against metabolic syndrome. The crystal structure of the complex between the phosphorylated-state mimic T172D mutant AMPK α2 kinase domain and a selective inhibitor, compound C, has been determined, revealing the unique inhibitor-binding mode of this protein kinase.
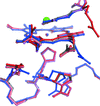
Crystal structures of midge larval haemoglobin under high salt concentrations at acidic pH were determined, which indicated that Cl− binding to haem iron results in a swinging movement of the distal Arg residue in this haemoglobin. Preliminary molecular-dynamics simulations of the haemoglobin also indicated that the movement of the Arg side chain was dependent on anionic ligand binding.
books received
Free








