journal menu
journal menu





issue contents
October 2000 issue
Low-resolution phasing
Proceedings of the CCP4 study weekend
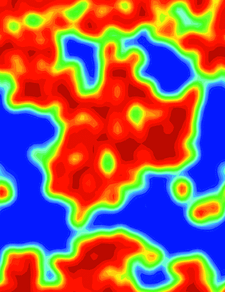
Cover illustration: Porcine pancreatic elastase, [100] section of the model envelope m(x) (p. 1316).




research papers
Open  access
access
access
This paper discusses how the phase problem in protein crystallography needs atomic resolution data for ab initio model-free phasing to work.
Open access
access
This paper reviews the contribution and future potential of cryo-electron microscopy and three-dimensional reconstruction in the determination of macromolecular structures.
Open access
access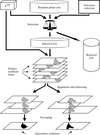
A low-resolution phasing statistical approach is presented and figures of merit which may be used successfully for phasing are discussed.
Open access
access
Applications of different density constraints in low-resolution phasing are discussed.
Open access
access
The possibility of using the statistical likelihood as a figure of merit in low-resolution ab initio phasing is tested.
Open access
access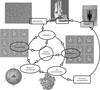
A description of the main steps of the structural analysis of non-crystalline biological macromolecules using cryo-electron microscopy is given in this paper.
Open access
access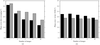
Electron crystallographic data from 2D crystals are being generated and published at an accelerating pace. However, unlike in X-ray crystallography, there are no standardized criteria in electron crystallography that can help investigators and reviewers alike to judge the reliability of electron crystallographic data. This article discusses some of the relevant issues and intends to serve as a platform for further discussion.
Open access
access
Analytical expressions are derived and computer simulations are used to assess the accuracy of common resolution measures for protein structures derived from images of single molecules of complexes. It is concluded that the alignment of images is accompanied by a correlation of noise, and that this correlation leads to erroneous resolution measurements.
Open access
access
The complementarity of X-ray and cryo-electron microscopy results is illustrated in the determination and interpretation of the structure of an enveloped virus at 9 Å resolution.
Open access
access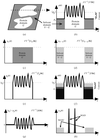
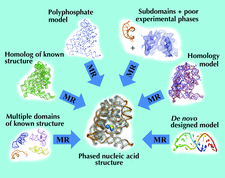
A complete account of the MASC method is given from the basic theory and experimental considerations through to the phasing of the macromolecular envelope at low resolution.
Open access
access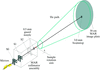
Modifications to a standard in-house X-ray diffraction camera (Elliot-GX13/300 mm MAR IP) are presented which allow the measurement of very low resolution diffraction intensities (>200 Å).
Open access
access
Sampling the diffraction pattern of a finite specimen more finely than the Nyquist frequency corresponds to surrounding the electron density of the specimen with a no-density region. This no-density region can be used to retrieve the phase information directly from the diffraction pattern.


Open access
access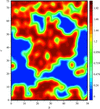
Homographic exponential models give a compact representation of probability distributions for the missing part of a macromolecular crystal structure and for the bulk solvent. These distributions are used in the maximum-likelihood refinement and maximum-entropy completion of partial structures.
PDB reference: porcine pancreatic elastase, 1lvy
Open access
access
Predicted values for wavelet coefficients are used to extend phases from 10 Å to around 6–7 Å.
Open access
access
Atomic models of large macromolecular complexes can be built by docking the atomic structures of components into cryo-EM maps. A procedure based on local real-space correlation is presented and verified by docking the domains from the crystal structure of GroEL into a cryo-EM map of the oligomer.
Open access
access
Techniques are described and examples are given for the interpretation of lower resolution cryo-EM results by using higher, near atomic resolution crystallographic results of component structures.
Open access
access
Two techniques of fitting X-ray to EM data in the presence of steric hindrance are presented and applied to a rotavirus–Fab complex. As the X-ray structures of the Fab and the Fab bound to a viral protein are known, this is a good test case.