

 journal menu
journal menu




issue contents
April 2025 issue
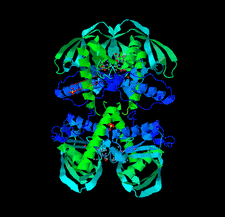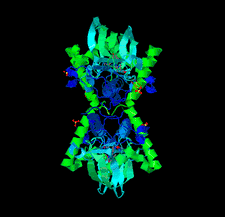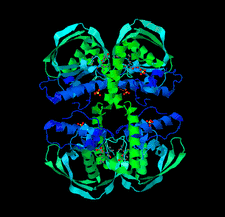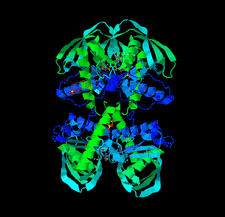
Cover illustration: Legionella pneumophila serogroup 1 peptide deformylase (PDF) inhibited by actinonin and Zn2+ crystallized as a tetramer. The actinonin molecules bind across PDF monomers [Nguyen et al. (2025), Acta Cryst. F81, 163–170].
research communications





Open  access
access
access
The 1.75 Å resolution structure of the G. haemolysans M26 IgA1 protease trypsin-like domain is presented. The structural data suggest that the domain exists in an inactive pro-enzyme-like state when in the context of the full-length protein. This putative pro-enzyme may be activated after being N-terminally excised from the larger M26 enzyme structure through the potential stabilization of its S1 pocket and rearrangement of adjacent surface loops.
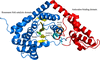
Crystal structures of tryptophanyl-tRNA synthetase from N. gonorrhoeae were solved in both the apo form and in complex with tryptophan to resolutions of 2.25 and 2.5 Å, respectively. These structures reveal conserved catalytic motifs and conformational changes at the active site upon ligand binding. Additionally, structural comparisons suggest that indolmycin may act as a competitive inhibitor, offering potential for antibiotic development.
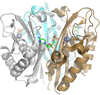
The co-crystal structures of five sulfonamide ligands bound to the active site of the IspF enzyme from B. psuedomallei are reported. The ligands include the drugs ethoxzolamide, acetazolamide, sulfapyridine and sulfamonomethoxine.

Download citation


Download citation
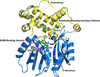
The crystal structure of UmaA from M. tuberculosis was solved to 1.95 Å resolution in space group P3221.
PDB reference: UmaA from Mycobacterium tuberculosis, 7lxi
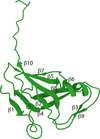
L. pneumophila serogroup 1 is the causative agent of Legionnaires' disease. Two crystal structures of apo and dUMP-bound deoxyuridine 5′-triphosphate nucleotidohydrolase (dUTPase) were determined to 1.80 and 1.95 Å resolution, respectively. dUTPases have been investigated as a potential druggable target in several pathogens.
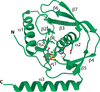
Peptide deformylases (PDFs) are of interest as viable drug targets for the development of new antimicrobials. Two crystal structures of PDF from L. pneumophila serogroup 1, the causative agent of Legionnaires' disease, bound to Ni2+ or to actinonin and Zn2+, were solved at 1.5 and 1.65 Å resolution, respectively.
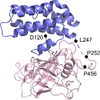
The purification, crystallization and determination of a 1.95 Å resolution structure of cyclophilin 37 from A. thaliana (AtCYP37) is reported. The structure, which is similar to those of Anabaena sp. CYPA and A. thaliana CYP38, is crucial for understanding how AtCYP37 interacts with the PetA subunit of cytochrome b6f, which is involved in the photoprotective mechanism of plants under high light conditions.
PDB reference: cyclophilin 37 from Arabidopsis thaliana, 9lh4
addenda and errata
Open access
access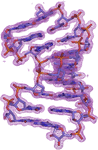
The first report of structural analysis of a nucleic acid using crystals grown in space. Corrigendum
The article by Ando et al. [(2025), Acta Cryst. F81, 95–100] is corrected.
![[publBio]](/logos/publbio.gif)





