
 journal menu
journal menu



issue contents
 | STRUCTURAL BIOLOGY |
ISSN: 2059-7983
CCP4 Study Weekend 2020
Model building, validation and representation in MX and cryoEM
Edited by Jon Agirre, Robbie Joosten and Alan Roseman
This virtual issue contains articles from the 2020 CCP4 Study Weekend.

Cover illustration: Structural Biology - The lost structure is a creative work that shows how solving a new structure is like an adventure, full of riddles and traps. The easier path never shows up the most amazing view of a landscape, like a revelation to human eyes for the first time. By Marco Salamina, Newcastle University.




Open  access
access
access
An introduction to the Proceedings of the 2020 CCP4 Study Weekend on model building which are available as a virtual issue at https://journals.iucr.org/special_issues/2020/CCP42020/.
Open access
access
A possible connection is discussed between hierarchical and other structures which maintain gender disparities in science and two instances where barriers to collaboration have hindered scientific progress.
Open access
access
The prerequisites for density modification of maps from electron cryomicroscopy are examined and a procedure for incorporating model-based information is presented.

Open access
access
A procedure for the identification of a protein in a map from electron cryomicroscopy based on automated model building and sequence assignment is presented.
Open access
access
This study shows the usefulness of integrating automated crystallographic model-building pipelines. We ran the four most used pipelines (ARP/wARP, Buccaneer, Phenix AutoBuild and SHELXE) alone and in pairwise combinations, and compared the structures that they produced based on structure completeness and Rfree.
Open access
access
Two neural networks were trained to predict the correctness of protein residues by combining multiple validation metrics in Coot. Using the predicted correctness to automatically prune models led to significant improvements in the Buccaneer pipeline.
Open access
access
Atomic models derived from cryo-EM data with map resolutions of better than 5 Å were automatically re-refined. The results of the computations are publicly available on a web page.
Open access
access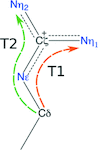
The geometry of arginine shows more complexity than is accommodated by the standard restraints.

Open access
access
New forms of adaptive or top-out distance and torsion restraints are described that are suitable for restraining a model to match a reference structure during interactive rebuilding. In addition, their implementation in ISOLDE is described, along with some illustrative example applications.
Open access
access
Virtual reality-specific tools for model building are possible, and can provide an order-of-magnitude speedup over mouse-and-keyboard tools in certain situations.
Open access
access
The mechanism for modelling covalent linkages in CCP4 is reviewed and the method of link-dictionary generation used by AceDRG is described. An overview of the various protocols available for the modelling and application of covalent linkages within the CCP4 suite is presented, providing instructive guidelines with a focus on practical application.
Open access
access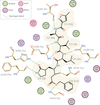
Analysis of the Protein Data Bank revealed that over a third of entries contain covalent linkages without descriptions in the CCP4 Monomer Library (CCP4-ML). The CCP4-ML was updated with AceDRG dictionaries corresponding to commonly occurring classes of missing linkages.
Open access
access
TEMPy2, an update of the TEMPy package to process, optimize and assess cryo-EM maps and the structures fitted to them, is presented.
Open access
access
Several approaches are presented to improve the performance of local correlation algorithms based on prior information about 3D search and target maps.
Open access
access
Reparametrizing the positions and flexibility of protein models for refinement against diffraction data results in a substantial reduction in the number of parameters. This produces maps that are less affected by model bias, which allow the maps to more faithfully reflect the true crystal contents and reveal more biological information.
Open access
access
The predictive power of simulation has become embedded in the infrastructure of modern economies. The scientific and technical progress that needs to be made to improve the predictive power of biomolecular simulations and how this might be achieved are discussed.
Open access
access
Methods for the validation of 3D maps from cryo-EM are described and illustrated using an example map of bacteriophage P22. Methods for fitting, flexible fitting, refinement and annotation of models generated based on such maps are also explored.
Open access
access
The dnatco.datmos.org web server performs an assignment of nucleic acid conformations and presents the results for the intuitive annotation, validation, modeling and refinement of nucleic acids.
Open access
access
Macromolecular atomic B-value distributions have been modelled using a mixture of shifted inverse-gamma distributions. B-value and resolution-dependent local ADP differences have also been applied for the validation of heavy atoms and ligands.
Open access
access
Visualization renders structural molecular data accessible to a broad audience. An approach to share molecular-visualization experiences based on FAIR principles is described. The workflow is exemplified with recent COVID-19-related data.
Open access
access
The LAHMA web server for structural analysis of homologous proteins is presented.
Open access
access
This article provides a survey of available web services and tools for data delivery and visualization of macromolecular structures.
Open access
access







