









 menu
menu

Navigation

- research papers
- research letters
- research papers
- scientific commentaries
- research papers
- advances
- research papers
- research papers
- scientific commentaries
- topical reviews
- research papers
- scientific commentaries
- research papers
- foundations
- research papers
- research papers
- advances
- research papers
- research papers
- scientific commentaries
- topical reviews
- research papers
issue contents
Focused issue on Quantum Crystallography
Edited by Paulina Dominiak, Angel Martín Pendás and Krzysztof Woźniak
This focused issue presents articles on Quantum Crystallography from various IUCr journals in celebration of the 100th anniversary of the development of Quantum Mechanics. Articles will be added to the issue during 2025.



Open  access
access
access
Recent methodological developments and their applications in quantum crystallography are reviewed, with an eye towards near-future advancements in this research field.

Download citation



Download citation
Kinematical Hirshfeld atom refinement has been applied to electron diffraction data for the first time, but the effect of using an aspherical atom model is overshadowed by dynamical scattering effects. Dynamical independent atom model refinement leads to significantly improved structures, suggesting that dynamical refinement is also necessary to obtain the full advantage of using aspherical atom models.
By implementing the aspherical atom model to normal mode refinement, we obtained accurate structures [including H-atom positions and anisotropic displacement parameters (ADPs)] and heat capacity from single-crystal X-ray diffraction data.

Understanding dynamic processes in molecular crystals is becoming crucial for the development of next-generation smart crystalline materials. In this context, Zwolenik & Makal [(2025). IUCrJ, 12, 23–35] shed light on the complex dynamics–structure–properties relation of a pyrene derivative by correlating molecular and lattice anharmonic vibrations with the unusual thermal expansion of the compound.
Download citation

Download citation
A recently discovered β polymorph of 1,3-diacetylpyrene has turned out to be a prominent negative thermal expansion material. Its unique properties can be linked to anharmonic oscillations in the crystal structure. The onset and development of anharmonic behavior have been successfully tracked over a wide temperature range by single-crystal X-ray diffraction experiments. Sufficient diffraction data quality combined with modern quantum crystallography tools allowed a thorough analysis of the elusive anharmonic effects for a moderate-scattering purely organic compound.
We demonstrate that applying the alternative electron density partition in a Hirshfeld atom refinement may significantly improve the accuracy of hydrogen atom parameters. The new partition leads to less overlapping atomic densities. As a result, hydrogen atom parameters are less dependent on the structural parameters of their neighbours and their inaccuracies.
CCDC references: 2407692; 2407693; 2407694; 2407695; 2407696; 2407697; 2407698; 2407699; 2407700; 2407701; 2407702; 2407703; 2407704; 2407705; 2407706; 2407707; 2407708; 2407709; 2407710; 2407711; 2407712; 2407718; 2407719; 2407720; 2407721; 2407722; 2407723; 2407724; 2407725; 2407726; 2407727; 2407728; 2407729; 2407730; 2407731; 2407732; 2407733; 2407734; 2407735; 2407736; 2407737; 2407738; 2407739; 2407740; 2407741; 2407742; 2407743; 2407744; 2407745; 2407746; 2407747; 2407748; 2407749; 2407750; 2407751; 2407752; 2407753; 2407754; 2407755; 2407756; 2407757; 2407758; 2407759; 2407760; 2407761; 2407762; 2407763; 2407764; 2407765; 2407766; 2407774; 2407775; 2407776; 2407777; 2407778; 2407779; 2407780; 2407781; 2407782; 2407783; 2407784; 2407785; 2407786; 2407787; 2407949; 2407950; 2407951; 2407952; 2407953; 2407954; 2407955; 2407956; 2407957; 2407958; 2407959; 2407960; 2407961; 2407962; 2407963; 2407964; 2407965; 2407966; 2407967; 2407968; 2407969; 2407970; 2407971; 2407972; 2407973; 2407974; 2407975; 2407976; 2407977; 2407978; 2407979; 2407980; 2407981; 2407982; 2407983; 2407984; 2407985; 2407986; 2407987; 2407988; 2407989; 2407990; 2407991; 2407992; 2407993; 2408748; 2408749; 2408750; 2408751; 2408752; 2408753; 2408754
Open access
access
The next-generation machine-learning force field FFLUX is applied to ice polymorphs Ih, II and XV. Under the quasi-harmonic approximation, Gibbs free energies are calculated using FFLUX at a significantly reduced computational cost compared with the commonly used density functional theory methods. However, the parametrized non-bonded potentials negatively affect the accuracy of the model, leading to large errors in the free energies calculated.
The methodology proposed combines the DFT calculations and photoelasticity caused by uniaxial compression of the crystal lattice, with particular emphasis on its anisotropy. It can be considered as part of optical engineering aimed at preliminary assessment of the photoelastic properties of crystal materials, thus assisting in their selection for synthesis and relevant applications.
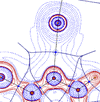
Interacting quantum atoms and source function studies on a series of halogen-bonded complexes between substituted pyridines and X2 or XCN molecules (X = I, Br) focus on the combined role played by the X and N interacting pairs and their local environment.

This study employs quantum crystallography to elucidate the selectivity of nonsteroidal anti-inflammatory drugs (NSAIDs), including ibuprofen, flurbiprofen, meloxicam and celecoxib, for cyclooxygenase-1 and cyclooxygenase-2 enzymes by analyzing binding energy and electrostatic interactions. The findings reveal key structural determinants of NSAID selectivity, providing valuable insights for the rational design of safer and more effective anti-inflammatory drugs.
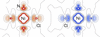
We analyse both charge and spin degrees of freedom in the electron density of NiX2(3,5-lutidine)4 (X = Cl, Br and I) to understand the nature of magnetic exchange via through-space Ni2+–halide⋯halide–Ni2+ interactions. We propose that remarkably strong interactions can occur in coordination polymers when the exchange pathway is `switched-on' by just very weak covalency and enhanced by a charge density that is naturally localized on the ligand's atoms that bond to the magnetic metal ions.
Free
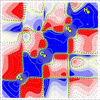
Yu & Gillet [Acta Cryst. (2025), B81, 168–180] extend quantum crystallography (QCr) by integrating real-space (1-RDM) and phase-space (Wigner function) representations, combining elastic and inelastic X-ray scattering to experimentally probe electron behavior in crystals. Their reconstruction of the quantum properties of a urea crystal bridges quantum chemistry and crystallography in a novel and imaginative way.
Open access
access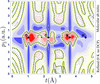
One hundred years after the quantum theory established position and momentum as incompatible quantities, quantum crystallography offers a way to visualize electron phase space behaviour in crystals.
Download citation
Download citation
Open access
access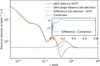
A procedure is presented for the use of effective core potentials during wavefunction-based refinements. The refinement quality of the results is indistinguishable from that of all-electron calculations, with speedups of a factor of 2 for compounds containing heavy elements.

The electron density and bonding nature of simple cubic Ca up to a phase 40 GPa is analyzed using theoretical quantum crystallography methods. The analysis is concerned with the electronic structure, pressure-induced electronic transitions, multi-center bond formation, topology of the electron density and atomic electronegativities.

The development of quantum crystallography depends on the availability of reliable theoretical electron densities. This work demonstrates a non-negligible code dependence of these densities and warns against their blind use.
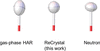
This work presents a new iterative refinement method, comparable to Hirshfeld atom refinement, using the Hansen–Coppens multipole model charge density description to obtain accurate atomic coordinates and atomic displacements based on CRYSTAL17 periodic boundary calculations. The refinement, performed using the Python code ReCrystal, allows the user to explore the full periodic charge density in the crystalline solid state for charge density analysis of weak interactions.
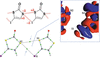
The design of crystalline solids relies on understanding and controlling intermolecular and intramolecular interactions. Through theoretical charge density analysis and database mining, Shukla et al. [(2025). IUCrJ, 12, 334–357] have found a new way of viewing supramolecular assembly through the lens of electrostatic complementarity.
Download citation
Download citation
This study establishes that hydrogen-, halogen- and chalcogen-bonding intermolecular and non-covalent intramolecular interactions are driven by a face-to-face orientation of electrophilic (charge-depleted) and nucleophilic (charge-concentrated) regions, which is the origin of the specific geometries found in synthons and supramolecular motifs.
CCDC reference: 2424586
Download citation
Download citation
Open access
access
The synthesis and quantum crystallographic analysis of the chemical bonding within WYLID, 2-(dimethyl-λ4-sulfaneylidene)-[1,2′-biindenylidene]-1′,3,3′(2H)-trione, a condensation product of YLID which is the most widely used calibrant for laboratory diffractometers, is presented. An ylid-type description of the S—C bond and a carbonyl-type description of the C—O bonds in WYLID is found in all aspects of the analysis.
Open access
access
This work explores the prediction of glucagon's dipole moments and polarizabilities using the GruPol database, incorporating ionic effects and solvation conditions. The results highlight the influence of high ionic concentrations on the protein's electrical properties, demonstrating good agreement with quantum mechanical benchmarks.
Open access
access
An approach is presented to calculate topological coordination numbers (tCNs) obeying the principle of coordination reciprocity from solid angles subtended by the interatomic surfaces of electron density (QTAIM) atomic domains. The tCN approach characterizes a compound's coordination situation as a set of sub-coordination scenarios with associated weights, which is considered a suitable input for future AI applications on structure–property relationships.
Download citation
Download citation
The temperature dependence of accurate structure factors of L-alanine and taurine was measured at the SPring-8 BL02B1 beamline. The quality of the structure factors is evaluated by charge density and quantum crystallographic studies. The effects of small amounts of twinning on the charge density study for taurine are also described.
Download citation
Download citation

This study reveals how additives and microwave radiation influence the crystallization of new tyramine polymorphs and their cocrystallization with barbital. The findings provide insights into polymorph stability and offer potential applications in molecular encapsulation and optical materials.
Download citation
Download citation
Open access
access
Multipolar model and Hirshfeld atom refinement are conducted on tetraaquabis(hydrogenmaleato)iron(II) based on a high-resolution X-ray diffraction experiment. Topological analysis is performed on both models. The study evaluates these models by comparing their effectiveness in determining bond lengths, anisotropic displacement parameters, electron densities, and atomic charges.
Open access
access
Measurement of diffuse scattering data and subsequent three-dimensional difference pair-distribution function (3D-ΔPDF) analysis of correlated disorder are compared for in-house and synchrotron sources.
Download citation
Download citation

The effect of a transition metal on the structural phase transition in 2D layered hybrid metal halides is reported.

The refinement flexibility of the Hansen–Coppens multipole model is tested on DFT calculated structure factors for the tetrakis(μ-acetato)diaquadicopper model system. The Cu scattering factor performs the best of all the options tried for most of the monitored parameters despite the Cu2+ nature of the complex studied. The Hansen–Coppens model performs similarly well when comparing deviations among computational chemistry methods.
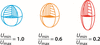
Combining improved diffraction methods, modeling approaches and advanced computations allows for a detailed understanding of atomic thermal motions in crystals. Thus, the Topical Review by Hoser & Madsen covers the Debye–Waller factor, the importance of anisotropic displacement parameters, and the interplay of experiment and theory to accurately capture collective atomic vibrations in molecular crystals.

This review commemorates the centenary of the Debye–Waller factor, highlighting its significance in quantifying the impact of thermal vibrations on scattering intensities as well as on crystal properties in small-molecule crystallography. It provides an introduction to thermal motion and displacement parameters, offering insights for chemists and crystallographers.
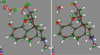
Single-crystal X-ray structures measured at around 20 K to high resolution were refined with structure-specific restraints from quantum chemical molecule-in-cluster and full-periodic computations, which permits benchmarking levels of theory of varying sophistication. Restraints can then `augment' low-quality crystal structures, with other possible applications.






